BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING
AND USING THE SAME
RELATED APPLICATION
This application claims the benefit of and priority to U.S. Patent Application No. 60/591,771, filed July 28, 2004, the disclosure of which is incorporated by reference herein.
FIELD OF THE INVENTION The present invention relates generally to the field of anti-infective, anti-pro liferative, anti-inflammatory, and prokinetic agents. More particularly, the invention relates to a family of biaryl heterocyclic compounds that are useful as therapeutic agents.
BACKGROUND
Since the discovery of penicillin in the 1920s and streptomycin in the 1940s, many new compounds have been discovered or specifically designed for use as antibiotic agents. It was once believed that infectious diseases could be completely controlled or eradicated with the use of such therapeutic agents. However, such beliefs have been shaken by the fact that strains of cells or microorganisms resistant to currently effective therapeutic agents continue to evolve. In fact, virtually every antibiotic agent developed for clinical use has ultimately encountered problems with the emergence of resistant- bacteria. For example, resistant strains of Gram- positive bacteria such as methicillin-resistant staphylocci, penicillin-resistant streptococci, and vancomycin-resistant enterococci have developed, which can cause serious and even fatal results for patients infected with such resistant bacteria. Bacteria that are resistant to macrolide antibiotics, i.e., antibiotics based on a 14- to 16-membered lactone ring, have developed. Also, resistant strains of Gram-negative bacteria such as H. influenzae and M. catarrhalis have been identified. See, e.g., F.D. Lowry, "Antimicrobial Resistance: The Example of Staphylococcus aureus " J. Clin. Invest, vol. I ll, no. 9, pp. 1265-1273 (2003); and Gold, H.S. and Moellering, R.C., Jr., "Antimicrobial-Drug Resistance," N. Engl. J. Med., vol. 335, pp. 1445-1453 (1996).
The problem of resistance is not limited to the area of anti-infective agents, because resistance has also been encountered with antiproliferative agents used in cancer chemotherapy. Therefore, there exists a need for new anti-infective and antiproliferative agents that are both effective against resistant bacteria and resistant strains of cancer cells. In the antibiotic area, despite the problem of increasing antibiotic resistance, no new major classes of antibiotics have been developed for clinical use since the approval in the United States in 2000 of the oxazolidinone ring-containing antibiotic, N-[[(5S)-3-[3-fluoro-4-(4- morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl acetamide, which is known as linezolid and is sold under the tradename Zyvox® (see compound A). See, R.C. Moellering, Jr., "Linezolid: The First Oxazolidinone Antimicrobial," Annals of Internal Medicine, vol. 138, no. 2, pp. 135-142 (2003).
Linezolid was approved for use as an anti-bacterial agent active against Gram-positive organisms. Unfortunately, linezolid-resistant strains of organisms are already being reported. See, Tsiodras et al., Lancet, vol. 358, p. 207 (2001); Gonzales et al, Lancet, vol. 357, p. 1179 (2001); Zurenko et al., Proceedings Of The 39* l Annual Inter science Conference On Antibacterial Agents And Chemotherapy (ICAAC) , San Francisco, CA, USA (September 26-29, 1999). Because linezolid is both a clinically effective and commercially significant anti¬ microbial agent, investigators have been working to develop other effective linezolid derivatives. Notwithstanding the foregoing, there is an ongoing need for new anti-infective and anti¬ proliferative agents. Furthermore, because many anti-infective and anti-proliferative agents have utility as anti-inflammatory agents and prokinetic agents, there is also an ongoing need for new compounds useful as anti-inflammatory and prokinetic agents.
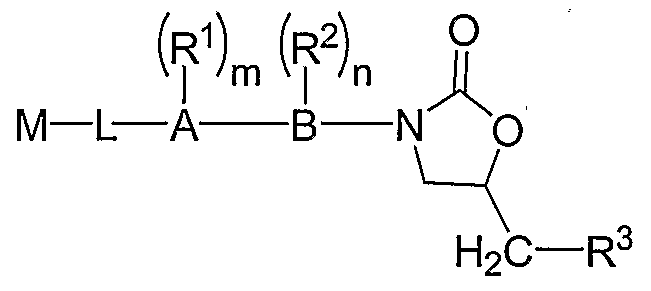
SUMMARY OF THE INVENTION The invention provides a family of compounds useful as anti-infective agents and/or anti¬ proliferative agents, for example, chemotherapeutic agents, anti-microbial agents, anti-bacterial
agents, anti-fungal agents, anti-parasitic agents, anti-viral agents, anti-inflammatory agents, and/or prokinetic (gastrointestinal modulatory) agents, having the formula:
(ψ )m (f )π M — L — A B— Het-CH2— R3 or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein A and B are selected from the group consisting of phenyl, pyridyl, pyrazinyl, pyrimidyl, and pyridazinyl; Het-CH2-R3 is selected from the group consisting of:
M is selected from the group consisting of: a) -CN, b) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each substituted with one or more -CN groups, c) optionally substituted C2-6 alkenyl or C2-6 alkynyl, or d) C1-6 alkyl, C2-6 alkenyl or G2-6 alkynyl, each with one or more carbons replaced with S(O)P; M-L is selected from the group consisting of M-X, M-X-L2, M-X-L^X-L2, and M-X-L2-X, or alternatively, L in M-L is a bond; and the variables L1, L2, R1, R2, R3, X, m, n, and p can be selected from the respective groups of chemical moieties or integers later defined in the detailed description. Particular embodiments of compounds of the invention include those having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein A, L, M, R
1, R
3, and m are selected from the respective groups of chemical moieties or integers later defined in the detailed description.
In addition, the invention provides methods of synthesizing the foregoing compounds. Following synthesis, an effective amount of one or more of the compounds can be formulated with a pharmaceutically acceptable carrier for administration to a mammal for use as an anti¬ cancer, anti-microbial, anti-biotic, anti-fungal, anti-parasitic or anti-viral agent, or to treat a proliferative disease, an inflammatory disease or a gastrointestinal motility disorder. The compounds or formulations can be administered, for example, via oral, parenteral, or topical routes, to provide an effective amount of the compound to the mammal.
The foregoing and other aspects and embodiments of the invention can be more fully understood by reference to the following detailed description and claims.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides a family of compounds that can be used as anti- proliferative agents and/or anti-infective agents. The compounds can be used without limitation, for example, as anti-cancer, anti-microbial, anti-bacterial, anti-fungal, anti-parasitic and/or anti¬ viral agents. Further, the present invention provides a family of compounds that can be used without limitation as anti-inflammatory agents, for example, for use in treating chronic inflammatory airway diseases, and/or as prokinetic agents, for example, for use in treating gastrointestinal motility disorders such as gastroesophageal reflux disease, gastroparesis (diabetic and post surgical), irritable bowel syndrome, and constipation.
1. Definitions
The term "substituted," as used herein, means that any one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency is not exceeded, and that the substitution results in a stable compound. When a substituent is keto (i.e., =0), then 2 hydrogens on the atom are replaced. Ring double bonds, as used herein, are double bonds that are formed between two adjacent ring atoms (e.g., C=C, C=N, or N=N).
The present invention is intended to include all isotopes of atoms occurring in the present compounds. Isotopes include those atoms having the same atomic number but different mass
numbers. By way of general example and without limitation, isotopes of hydrogen include tritium and deuterium, and isotopes of carbon include C- 13 and C- 14.
The compounds described herein can have asymmetric centers. Compounds of the present invention containing an asymmetrically substituted atom can be isolated in optically active or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of racemic forms or by synthesis from optically active starting materials. Many geometric isomers of olefins, C=N double bonds, and the like can also be present in the compounds described herein, and all such stable isomers are contemplated in the present invention. Cis and trans geometric isomers of the compounds of the present invention are described and can be isolated as a mixture of isomers or as separated isomeric forms. All chiral, diastereomeric, racemic, and geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated. All processes used to prepare compounds of the present invention and intermediates made therein are considered to be part of the present invention. All tautomers of shown or described compounds are also considered to be part of the present invention.
When any variable (e.g., R1) occurs more than one time in any constituent or formula for a compound, its definition at each occurrence is independent of its definition at every other occurrence. Thus, for example, if a group is shown to be substituted with 0-2 R1 moieties, then the group can optionally be substituted with up to two R1 moieties and R1 at each occurrence is selected independently from the definition of R1. Also, combinations of substituents and/or variables are permissible, but only if such combinations result in stable compounds.
When a bond to a substituent is shown to cross a bond connecting two atoms in a ring, then such substituent can be bonded to any atom in the ring. When a substituent is listed without indicating the atom via which such substituent is bonded to the rest of the compound of a given formula, then such substituent can be bonded via any atom in such substituent. Combinations of substituents and/or variables are permissible, but only if such combinations result in stable compounds.
Compounds of the present invention that contain one or more nitrogen atoms can be converted to N-oxides by treatment with an oxidizing agent (e.g., metα-chloroperoxybenzoic acid (m-CPBA) and/or hydrogen peroxide) to afford other compounds of the present invention. Thus, all shown and claimed nitrogen-containing compounds are considered, when allowed by valency and structure, to include both the compound as shown and its N-oxide derivative (which
can be designated as N->0 or N+-O"). Furthermore, in other instances, the nitrogen atom(s) in the compounds of the present invention can be converted to N-hydroxy or N-alkoxy compounds. For example, N-hydroxy compounds can be prepared by oxidation of the parent amine by an oxidizing agent such as m-CPBA. All shown and claimed nitrogen-containing compounds are also considered, when allowed by valency and structure, to cover both the compound as shown and its N-hydroxy (i.e., N-OH) and N-alkoxy (i.e., N-OR, wherein R is substituted or unsubstituted C1-6 alkyl, alkenyl, alkynyl, C3-14 carbocycle, or 3-14-membered heterocycle) derivatives.
Similarly, compounds of the present invention that contain one or more sulfur atoms can be converted to S-oxides, i.e., sulfoxides or sulfones, by treatment with an oxidizing agent (e.g., m-CPBA and/or hydrogen peroxide) to afford other compounds of the present invention. Thus, all shown and claimed sulfur-containing compounds are considered, when allowed by valency and structure, to include both the compound as shown and its S-oxide derivatives (which can be designated as S(O)P, where p = 1 or 2). When an atom or a chemical moiety is followed by a subscripted numeric range (e.g., C1-
6), the invention is meant to encompass each number within the range as well as all intermediate ranges. For example, "C1-6 alkyl" is meant to include alkyl groups with 1, 2, 3, 4, 5, 6, 1-6, 1-5, 1-4, 1-3, 1-2, 2-6, 2-5, 2-4, 2-3, 3-6, 3-5, 3-4, 4-6, 4-5, and 5-6 carbons.
As used herein, "alkyl" is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms. For example, Cμg alkyl is intended to include Ci, C2, C3, C4, C5, and Ce alkyl groups. Examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl, t-butyl, n-pentyl, s-pentyl, and n-hexyl.
As used herein, "alkenyl" is intended to include hydrocarbon chains of either straight or branched configuration having one or more carbon-carbon double bonds occurring at any stable point along the chain. For example, C2-6 alkenyl is intended to include C2, C3, C4, C5, and Ce alkenyl groups. Examples of alkenyl include, but are not limited to, ethenyl and propenyl.
As used herein, "alkynyl" is intended to include hydrocarbon chains of either straight or branched configuration having one or more carbon-carbon triple bonds occurring at any stable point along the chain. For example, C2-6 alkynyl is intended to include C2, C3, C4, C5, and Ce alkynyl groups. Examples of alkynyl include, but are not limited to, ethynyl and propynyl.
Furthermore, "alkyl", "alkenyl", and "alkynyl" are intended to include moieties which are diradicals, i.e., having two points of attachment, an example of which in the present invention is when L is selected from these chemical groups. A nonlimiting example of such an alkyl moiety that is a diradical is -CH2CH2-, i.e., a C2 alkyl group that is covalently bonded via each terminal carbon atom to the remainder of the molecule.
As used herein, "halo" or "halogen" refers to fluoro, chloro, bromo, and iodo. "Counterion" is used to represent a small, negatively charged species such as fluoride, chloride, bromide, iodide, hydroxide, acetate, and sulfate.
As used herein, "carbocycle" or "carbocyclic ring" is intended to mean any stable monocyclic, bicyclic, tricyclic, or higher order cyclic ring having the specified number of carbons, any of which can be saturated, unsaturated, or aromatic, recognizing that rings with certain numbers of members cannot be bicyclic or tricyclic, e.g., a 3-membered ring can only be a monocyclic ring. For example, a C3-14 carbocycle is intended to mean a monocyclic, bicyclic, tricyclic, or higher order cyclic ring having 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, or 14 carbon atoms. Examples of carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, cyclooctyl, cyclooctenyl, cyclooctadienyl, adamantly, fluorenyl, phenyl, naphthyl, indanyl, anthryl, phenanthryl, and tetrahydronaphthyl. Bridged rings are also included in the definition of carbocycle, including, for example, [3.3.0]b^cyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane, and [2.2.2]bicyclooctane. A bridged ring occurs when one or more carbon atoms link two non-adjacent carbon atoms. Preferred bridges are one or two carbon atoms. It is noted that a bridge always converts a monocyclic ring into a tricyclic ring. When a ring is bridged, the substituents recited for the ring can also be present on the bridge. Fused (e.g., naphthyl and tetrahydronaphthyl) and spiro rings are also included. It should be understood that included in the definition of "carbocycle" and "carbocyclic ring" are "aromatic carbocycles" and "aromatic carbocyclic rings," which are "aryl" groups. In the case of bicyclic aromatic carbocyclic rings, only one of the rings needs to be aromatic (e.g., tetrahydronaphthyl), though both can be (e.g., naphthyl). Similarly, in the case of tricyclic or higher order aromatic carbocyclic rings, only one of the rings needs to be aromatic, although tricyclic or higher order aromatic carbocycles having more than one aromatic ring are included (e.g., fluorenyl). Examples of aromatic carbocycles include, but are not limited to, phenyl,
naphthyl, tetrahydronaphthyl, indanyl, indenyl, phenanthryl, anthryl, fluorenyl, pentalenyl, azulyl, chrysyl, pyryl, tetracyl, fluranthyl, coronyl, and hexahelicyl.
As used herein, the term "heterocycle" or "heterocyclic" is intended to mean any stable monocyclic, bicyclic, tricyclic, or higher order cyclic ring (recognizing that rings with certain numbers of members cannot be bicyclic or tricyclic, e.g., a 3-membered ring can only be a monocyclic ring), which is saturated, unsaturated, or aromatic and comprises carbon atoms and one or more ring heteroatoms, e.g., 1 or 1-2 or 1-3 or 1-4 or 1-5 or 1-6 heteroatoms, independently selected from the group consisting of nitrogen, oxygen, and sulfur. A bicyclic or tricyclic heterocycle can have one or more heteroatoms located in one ring, or the heteroatoms can be located in more than one ring. The nitrogen and sulfur heteroatoms can optionally be oxidized (e.g., N→O and S(O)P, where p = 1 or 2). When a nitrogen atom is included in the ring it is either N or NH, depending on whether or not it is attached to a double bond in the ring (i.e., a hydrogen is present if needed to maintain the tri- valency of the nitrogen atom). The nitrogen atom can be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, as defined). The heterocyclic ring can be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure. The heterocyclic rings described herein can be substituted on carbon or on a nitrogen atom if the resulting compound is stable. A nitrogen hi the heterocycle can optionally be quaternized. It is preferred that when the total number of S and O atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another. Bridged rings are also included in the definition of heterocycle. A bridged ring occurs when one or more atoms (i.e., C, O, N, or S) link two non-adjacent carbon or nitrogen atoms. Preferred bridges include, but are not limited to, one carbon atom, two carbon atoms, one nitrogen atom, two nitrogen atoms, and a carbon-nitrogen group. It is noted that a bridge always converts a monocyclic ring into a tricyclic ring. When a ring is bridged, the substituents recited for the ring can also be present on the bridge. Spiro and fused rings are also included.
As used herein, the term "aromatic heterocycle" or "heteroaryl" is intended to mean a stable monocyclic, bicyclic, or higher order aromatic heterocyclic ring which consists of carbon atoms and one or more heteroatoms, e.g., 1 or 1-2 or 1-3 or 1-4 or 1-5 or 1-6 heteroatoms, independently selected from the group consisting of nitrogen, oxygen, and sulfur. For example, an aromatic heterocycle or heteroaryl can be a 5, 6, 7, 8, 9, 10, 11, or 12-membered monocyclic or bicyclic aromatic heterocyclic ring, recognizing that rings with certain numbers of members cannot be a bicyclic aromatic, e.g., a 5-membered ring can only be a monocyclic aromatic ring.
In the case of bicyclic heterocyclic aromatic rings, only one of the two rings needs to be aromatic (e.g., 2,3-dihydroindole), though both can be (e.g., quinoline). The second ring can also be fused or bridged as defined above for heterocycles. The nitrogen atom can be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, as defined). The nitrogen and sulfur heteroatoms can optionally be oxidized (i.e., N→O and S(O)P, where p = 1 or 2). It is to be noted that total number of S and O atoms in the aromatic heterocycle is generally not more than l.
Examples of heterocycles include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzoxazolinyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4ai/-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2/f,6H-l,5,2-dithiazinyl, dihydrofuro[2,3-&]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, lH-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, 3Η-indolyl, isatinoyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, methylenedioxyphenyl, morpholinyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3-oxadiazolyl, 1,2,4- oxadiazolyl, 1,2,5-oxadiazolyl, 1,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxindolyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, piperidonyl, 4-piperidonyl, piperonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, 2H-pyrrolyl, pyrrolyl, quinazolinyl, quinolinyl, 4H-quinolizinyl, quinoxalinyl, quinuclidinyl, tetrahydrofuranyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, tetrazolyl, 6H- 1,2,5- thiadiazinyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,2,5-thiadiazolyl, 1,3,4-thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1,2,3-triazolyl, 1,2,4-triazolyl, 1,2,5-triazolyl, 1,3,4-triazolyl, and xanthenyl.
As used herein, the phrase "pharmaceutically acceptable" refers to those compounds, materials, compositions, carriers, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
As used herein, "pharmaceutically acceptable salts" refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines, alkali or organic salts of acidic residues such as carboxylic acids, and the like. The pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include, but are not limited to, those derived from inorganic and organic acids selected from 2- acetoxybenzoic, 2-hydroxyethane sulfonic, acetic, ascorbic, benzene sulfonic, benzoic, bicarbonic, carbonic, citric, edetic, ethane disulfonic, ethane sulfonic, fumaric, glucoheptonic, gluconic, glutamic, glycolic, glycollyarsanilic, hexylresorcinic, hydrabamic, hydrobromic, hydrochloric, hydroiodic, hydroxymaleic, hydroxynaphthoic, isethionic, lactic, lactobionic, lauryl sulfonic, maleic, malic, mandelic, methane sulfonic, napsylic, nitric, oxalic, pamoic, pantothenic, phenylacetic, phosphoric, polygalacturonic, propionic, salicyclic, stearic, subacetic, succinic, sulfamic, sulfanilic, sulfuric, tannic, tartaric, and toluene sulfonic.
The pharmaceutically acceptable salts of the present invention can be synthesized from a parent compound that contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred.' Lists of suitable salts are found in Remington's Pharmaceutical Sciences, 18th ed. (Mack Publishing Company, 1990). For example, salts can include, but are not limited to, the hydrochloride and acetate salts of the aliphatic amine- containing, hydroxyl amine-containing, and imine-containing compounds of the present invention. A nonlimiting example of a salt of a compound of the present invention is the monohydrochloride salt of compound 133 as exemplified in Example 28.
Additionally, the compounds of the present invention, and particularly the salts of the compounds, can exist in either hydrated or unhydrated (the anhydrous) form or as solvates with other solvent molecules. Nonlimiting examples of hydrates include monohydrates, dihydrates, etc. Nonlimiting examples of solvates include ethanol solvates, acetone solvates, etc.
Since prodrugs are known to enhance numerous desirable qualities of pharmaceuticals (e.g., solubility, bioavailability, manufacturing, etc.) the compounds of the present invention can
be delivered in prodrug form. Thus, the present invention is intended to cover prodrugs of the presently claimed compounds, methods of delivering the same and compositions containing the same. "Prodrugs" are intended to include any covalently bonded carriers that release an active parent drug of the present invention in vivo when such prodrug is administered to a mammalian subject. Prodrugs the present invention are prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound. Prodrugs include compounds of the present invention wherein a hydroxy, amino, or sulfhydryl group is bonded to any group that, when the prodrug of the present invention is administered to a mammalian subject, cleaves to form a free hydroxyl, free amino, or free sulfhydryl group, respectively. Examples of prodrugs include, but are not limited to, acetate, formate, and benzoate derivatives of alcohol and amine functional groups in the compounds of the present invention.
"Stable compound" and "stable structure" are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
As used herein, "treating" or "treatment" means the treatment of a disease-state in a mammal, particularly in a human, and include: (a) preventing the disease-state from occurring in a mammal, in particular, when such mammal is predisposed to the disease-state but has not yet been diagnosed as having it; (b) inhibiting the disease-state, i.e., arresting its development; and/or (c) relieving the disease-state, i.e., causing regression of the disease state.
As used herein, "mammal" refers to human and non-human patients.
As used herein, the term "effective amount" refers to an amount of a compound, or a combination of compounds, of the present invention effective when administered alone or in combination as an antiproliferative and/or anti-infective agent. In particular, an effective amount refers to an amount of the compound present in or on a recipient sufficient to elicit biological activity, for example, anti-infective activity (e.g., anti-microbial activity, anti-fungal activity, anti-viral activity, anti-parasitic activity) and/or antiproliferative activity. The combination of compounds optionally is a synergistic combination. Synergy, as described, for example, by Chou and Talalay, Adv. Enzyme Regul., vol. 22, pp. 27-55 (1984), occurs when the effect of the compounds when administered in combination is greater than the additive effect of the compounds when administered alone as a single agent. In general, a synergistic effect is most clearly demonstrated at sub-optimal concentrations of the compounds. Synergy can be in
terms of lower cytotoxicity, increased antiproliferative and/or anti-infective effect, or some other beneficial effect of the combination compared with the individual components.
All percentages and ratios used herein, unless otherwise indicated, are by weight.
Throughout the description, where compositions are described as having, including, or comprising specific components, it is contemplated that compositions also consist essentially of, or consist of, the recited components. Similarly, where processes are described as having, including, or comprising specific process steps, the processes also consist essentially of, or consist of, the recited processing steps. Further, it should be understood that the order of steps or order for performing certain actions are immaterial so long as the invention remains operable. Moreover, two or more steps or actions can be conducted simultaneously.
2. Compounds of the Invention .
In one aspect, the invention provides compounds having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein: A is selected from the group consisting of: phenyl, pyridyl, pyrazinyl, pyrimidinyl, and pyridazinyl;
B is selected from the group consisting of: phenyl, pyridyl, pyrazinyl, pyrimidinyl, and pyridazinyl;
Het-CH2-R3 is selected from the group consisting of:
M is selected from the group consisting of: a) -CN; b) i) C1-6 alkyl, ii) C2-6 alkenyl, or iii) C2-6 alkynyl, wherein any of i) - iii) is substitued with one or more -CN groups and optionally is further substituted with one or more R4 groups,
c) i) C2-6 alkenyl or ii) C2-6 alkynyl, wherein any of i) - ii) optionally is substituted with one or more R4 groups, and d) i) C1-6 alkyl, ii) C2-6 alkenyl, or iii) C2-6 alkynyl, wherein one or more carbons of any of i) — iii) is replaced with
S(O)p and one or more of the remaining carbons optionally is further substituted with one or more R4 groups, provided that
(a) when a carbon bonded to L is replaced with S(O)P, then p is 0 or 1, and (b) ■ when one or more of the remaining carbons is substituted with one or more R4 groups, then R4 does not include =0 or =S;
M-L is selected from the group consisting of: a) M-X, b) M-X-L2, c) M-X-L'-X-L2, and d) M-X-L2-X, wherein X, at each occurrence,' independently is selected from the group consisting of: i) -O- ii) -NR4- iii) -N(O) - iv) -N(OR4) - v) -S(O)P- vi) -NR4-N= vii) =N-NR4-, viii) -0-N=, ix) =N-O- x) -N=, xi) =N, xii) -NR4-NR4-, xiii) -NR4-0- xiv) -0-NR4-, xv) -NR4C(O)O-, xvi) -OC(O)NR4- xvii) -NR4C(O)NR4-, xviii) -NR4C(NR4)NR4- xix)
xx)
L
1 is selected from the group consisting of: a) C
1-6 alkyl, b) C
2-6 alkenyl, and c) C
2-6 alkynyl, wherein any of a) - c) optionally is substituted with one or more R
4 groups; and
L2 is selected from the group consisting of: a) C1-6 alkyl, b) C2-6 alkenyl, and c) C2-6 alkynyl,
wherein any of a) - c) optionally is substituted with one or more R4 groups; alternatively, L in M-L is a bond;
R1, at each occurrence, independently is selected from the group consisting of: a) F, b) Cl5 c) Br, d) I5 e) -CF3, f) -OR7, g) -CN5 h) -NO2, i) -NR7R7, j) -C(O)R7, k) -C(O)OR7, 1) -OC(O)R7, m) -C(O)NR7R7, n) -NR7C(O)R7, o) -OC(O)NR7R7, p) -NR7C(O)OR7, q) -NR7C(O)NR7R7, r) -C(S)R7, s) -C(S)OR7, t) -OC(S)R7, u) -C(S)NR7R7, v) -NR7C(S)R7, w) -OC(S)NR7R7, x) -NR7C(S)OR7, y) -NR7C(S)NR7R7, z) -NR7C(NR7)NR7R7, aa) -S(O)PR7, bb) -SO2NR7R7, and cc) R7;
R2, at each occurrence, independently is selected from the group consisting of: a) F, b) Cl, c) Br, d) I, e) -CF3, f) -OR7, g) -CN5 h) -NO2, i) -NR7R7, j) -C(O)R7, k) -C(O)OR7, 1) -OC(O)R7, m) -C(O)NR7R7, n) -NR7C(O)R7, o) -OC(O)NR7R7, p) -NR7C(O)OR7, q) -NR7C(O)NR7R7, r) -C(S)R7, s) -C(S)OR7, t) -OC(S)R7, u) -C(S)NR7R7, v) -NR7C(S)R7, w) -OC(S)NR7R7, x) -NR7C(S)OR7, y) -NR7C(S)NR7R7, z) -NR7C(NR7)NR7R7, aa) -S(O)PR7, bb) -SO2NR7R7, and cc) R7; R3 is selected from the group consisting of: a) -OR7, b) -NR7R7, c) -C(O)R7, d) -C(O)OR7, e) -OC(O)R7, f) -C(O)NR7R7, g) -NR7C(O)R7, h) -OC(O)NR7R7, i) -NR7C(O)OR7, j) -NR7C(O)NR7R7, k) -C(S)R7, 1) -C(S)OR7, m) -OC(S)R7, n) -C(S)NR7R7, o) -NR7C(S)R7, p) -OC(S)NR7R7, q) -NR7C(S)OR7, r) -NR7C(S)NR7R7, s) -NR7C(NR7)NR7R7, t) -S(O)PR7, u) -SO2NR7R7, and v) R7;
R4, at each occurrence, independently is selected from the group consisting of: a) H, b) =0, c) =S, d) =NR5, e) =N0R5, f) =N-NR5R5, g) -OR5, h) -CN, i) -NO2, j) -NR5R5, k) -C(O)R5, 1) -C(O)OR5, m) -OC(O)R5, n) -C(O)NR5R5, o) -NR5C(O)R5, p) -OC(O)NR5R5, q) -NR5C(O)OR5, r) -NR5C(O)NR5R5, s) -C(S)R5, t) -C(S)OR5, u) -OC(S)R5, v) -C(S)NR5R5, w) -NR5C(S)R5, x) -OC(S)NR5R5, y) -NR5C(S)OR5, z) -NR5C(S)NR5R5, aa) -NR5C(NR5)NR5R5, bb) -S(O)PR5, and cc) R5;
R5, at each occurrence, independently is selected from the group consisting of: a) H, b) C1-6 alkyl, c) C2-6 alkenyl, d) C2-6 alkynyl, e) -C(O)-C1-6 alkyl,
f) -C(O) -C2-6 alkenyl, g) -C(O) -C2-6 alkynyl, h) -C(O)O-C1-6 alkyl, i) -C(O)O-C2-6 alkenyl, and j) -C(O)O-C2-6 alkynyl, wherein any of b) - j) optionally is substituted with one or more R6 groups; R6, at each occurrence, independently is selected from the group consisting of: a) -OH, b) -OCi-6 alkyl, c) -SH, d) -SC1-6 alkyl, e) -CN, f) -NO2, g) -NH2, h) -NHC1-6 alkyl, i) -N(C1-6 alkyl)2, j) -C(O)C1-6 alkyl, k) -C(O)OC1-6 alkyl, 1) -C(O)NH2, m) -C(O)NHC1-6 alkyl, n) -C(O)N(C1-6 alkyl)2, o) -NHC(O)C1-6 alkyl, and p) -S(O)pC1-6 alkyl; R7, at each occurrence, independently is selected from the group consisting of: a) H, b) C1-6 alkyl, c) C2-6 alkenyl, d) C2-6 alkynyl, e) C3-14 saturated, unsaturated, or aromatic carbocycle, f) 3-14 membered saturated, unsaturated, or aromatic heterocycle comprising one or more heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur, g) -C(O) -C1-6 alkyl, h) -C(O)-C2-6 alkenyl, i) -C(O)-C2-6 alkynyl, j) -C(O)-C3-14 saturated, unsaturated, or aromatic carbocycle, k) -C(O)-3-14 membered saturated, unsaturated, or aromatic heterocycle comprising one or more heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur, 1) -C(O)O-C1-6 alkyl, m) -C(O)O-C2-6 alkenyl, n) -C(O)O-C2-6 alkynyl, o) -C(O)O-C3-14 saturated, unsaturated, or aromatic carbocycle, and p) -C(O)O-3-14 membered saturated, unsaturated, or aromatic heterocycle comprising one or more heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur, wherein any of b) - p) optionally is substituted with one or more R8 groups; R8, at each occurrence, is independently selected from the group consisting of: a) F5 b) Cl, c) Br, d) I, e) =0, f) =S, g) =NR9, h) =NOR9, i) =N-NR9R9, j) -CF3, k) -OR9, 1) -CN, m) -NO2, n) -NR9R9, o) -C(O)R9, p) -C(O)OR9, q) -OC(O)R9, r) -C(O)NR9R9, s) -NR9C(O)R9, t) -OC(O)NR9R9, u) -NR9C(O)OR9, v) -NR9C(O)NR9R9, w) -C(S)R9, x) -C(S)OR9, y) -OC(S)R9, z) -C(S)NR9R9, aa) -NR9C(S)R9, bb) -OC(S)NR9R9, cc) -NR9C(S)OR9, dd) -NR9C(S)NR9R9, ee) -NR9C(NR9)NR9R9, ff) -S(O)PR9, gg) -SO2NR9R9, and hh) R9;
R9, at each occurrence, independently is selected from the group consisting of: a) H, b) C1-6 alkyl, c) C2-6 alkenyl, d) C2-6 alkynyl, e) C3-14 saturated, unsaturated, or aromatic carbocycle, f) 3-14 membered saturated, unsaturated, or aromatic heterocycle comprising one or more heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur, g) -C(O)-C1-6 alkyl, h) -C(O)-C2-6 alkenyl, i) -C(O)-C2-6 alkynyl, j) -C(O)-C3-14 saturated, unsaturated, or aromatic carbocycle, k) -C(O)-3-14 membered saturated, unsaturated, or aromatic heterocycle comprising one or more heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur, 1) -C(O)O-C1-6 alkyl, m) -C(O)O-C2-6 alkenyl, n) -C(O)O-C2-6 alkynyl, o) -C(O)O-C3-14 saturated, unsaturated, or aromatic carbocycle, and p) -C(O)O-3-14 membered saturated, unsaturated, or aromatic heterocycle comprising one or more heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur, wherein any of b) - p) optionally is substituted with one or more moieties selected from the group consisting of: i) F, ii) Cl, iii) Br, iv) I, v) -CF3, vi) -OH, vii) -OC1-6 alkyl, viii) -SH, ix) -SC1-6 alkyl, x) -CN, xi) -NO2, xii) -NH2, xiii) -NHCi-6 alkyl, xiv) -N(C1-6 alkyl)2, xv) -C(O)C1-6 alkyl, xvi) -C(O)OC1-6 alkyl, xvii) -C(O)NH2, xviii) -C(O)NHC1-6 alkyl, xix) -C(O)N(C1-6 alkyl)2, xx) -NHC(O)Ci-6 alkyl, xxi) -SO2NH2- xxii) -SO2NHCi-6 alkyl, xxiii) -SO2N(C1-6 alkyl)2, and xxiv) -S(O)pCi-6 alkyl; m is 0, 1, 2, 3, or 4; n is O, 1, 2, 3, or 4; and p, at each occurrence, independently is 0, 1, or 2; and wherein the compound does not have the formula selected from the group consisting of:
Particular embodiments of the invention include compounds having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein A, B, L, M, R
1, R
2, R
3, m, and n are defined above.
Other embodiments include compounds having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein A, B, L, M, R , R , R , m, and n are defined as described above.
Particular compounds include those where A and B independently are selected from the group consisting of phenyl and pyridyl, and m and n independently are O, 1, or 2.
In some embodiments, A-B is:
wherein A, R , and n are defined as described above. Particular compounds according to these embodiments include those where R
2 is selected from the group consisting of H and F, and n is 0, 1, or 2. In particular embodiments, A-B is:
wherein A is defined as described above.
In various embodiments, A-B is:
wherein B is defined as described in above.
In some embodiments, R3 is -NR7C(O)R7. In other embodiments, R3 is -NHC(O)R7. Particular compounds according to these embodiments include those where R7 is a C1-6 alkyl group optionally substituted with one or more substituents independently selected from F or Cl. Examples of such alkyl groups include, but are not limited to, -CH3, -CH2F, -CHF2, -CF3, -CH2Cl, -CHCl2, -CCl3, -CHFCl, -CF2Cl, and -CFCl2. In other embodiments, R3 is:
Particular embodiments of the invention include compounds having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein A, B, L, M, R
1, R
2, m, and n are defined as described above.
Other embodiments of the invention include compounds having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein L, M, R
1, R
3, and m are defined as described above, A is selected from the group consisting of phenyl and pyridyl, R
2 is selected from the group consisting of H and F, and n is 0, 1, or 2.
Still other embodiments of the invention include compounds having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein A, L, M, R
1, R
3, and m are defined as described above. Particular compounds according to these embodiments include compounds having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein A, L, M, R
1, and m are defined as described above.
Some embodiments of the invention include compounds having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein L, M, and R are defined as described above. Particular compounds according to these embodiments include those having the formula:
or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein L and M are defined as described above.
In some embodiments, L is a bond and M is -CN. In other embodiments, L is a bond and M is selected from the group consisting of:
OR4 a) C2-6 alkenyl, b) C2-6 alkynyl, c) NC-C1-6 alkyl-, d) NC-C2-6 alkenyl-, e) NC-CH-,
NR4R4 CHO CH2OR4 CH2NR4R4 OR4 f) NC-CH- , g) NC-CH- , h) NC-CH- , i) NC-CH- , j) NC-CH2-CH-, and
NR4R4 k) NC-CH2-CH-
Particular compounds according to these embodiments include those wherein M is selected from the group consisting of:
NH2 CH2OH a) NC-CH2-, b) NC-CH2CH2-, c) NC-CH-CH-, d) NC-CH-, e) NC-CH- ,
OH NH2 f) NC-CH2-CH-, and g) NC-CH2-CH-.
hi some embodiments, L is X-L2, and X is -NR4-. In these embodiments, R4 can be, for example, H, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, or -C(O)C1-6 alkyl. Particular compounds according to these embodiments include those where R4 is selected from the group consisting of: a) H, b) -CH3, c) -C(O)CH3, d) -CH2CH=CH2, e) -CH2CH2CH=CH2, f) -CH2CH2CH2CH=CH2, g) -CH2CH=CHCH3, h) -CH2C=CH, i) -CH2C=CCH3, and j) -CH2CH2CH2C=CH.
Compounds according to these embodiments include those where L2 is C1-6 alkyl and/or M is selected from the group consisting of C2-6 alkenyl, C2-6 alkynyl, NC-CH2CH2-, NC-CH2C(O)- and NC-CH2-. hi particular, M-L can be NC-CH2CH2NHCH2- NC-CH2C(O)NHCH2- or NC-CH2NHCH2-.
Still other embodiments include compounds where L is X-L2, and X is
In these embodiments, L can be C1-6 alkyl. Particular examples include compounds where M-L is
In another aspect, the invention provides methods for synthesizing compounds and pharmaceutically acceptable salts thereof according to the invention. In yet another aspect, the invention provides a pharmaceutical composition comprising an effective amount of one or more of the foregoing compounds and a pharmaceutically acceptable carrier. Suitable formulating agents are described in detail in section 4 hereinbelow.
One or more of the foregoing compounds can also be incorporated into a medical device. For example, a medical device, such as a medical stent, can contain or be coated with one or more of the compounds of the invention.
In still another aspect, the invention provides a method for treating a microbial infection, a fungal infection, a viral infection, a parasitic disease, a proliferative disease, an inflammatory disease, or a gastrointestinal motility disorder in a mammal. The method involves administering an effective amount of one or more compounds or pharmaceutical compositions of the invention, for example, via oral, parenteral or topical routes.
The invention provides a method of treating a disorder in a mammal comprising the step of administering to the mammal an effective amount of one or more compounds of the invention thereby to ameliorate a symptom of a particular disorder. Such a disorder can be selected from the group consisting of a skin infection, nosocomial pneumonia, post- viral pneumonia, an abdominal infection, a urinary tract infection, bacteremia, septicemia, endocarditis, an atrio¬ ventricular shunt infection, a vascular access infection, meningitis, surgical prophylaxis, a peritoneal infection, a bone infection, a joint infection, a rnethiciUin-resistant Staphylococcus aureus infection, a vancomycin-resistant Enterococci infection, a linezolid-resistant organism infection, and tuberculosis.
3. Characterization of Compounds of the Invention
Compounds designed, selected and/or optimized by methods described above, after being produced, can be characterized using a variety of assays known to those skilled in the art to determine whether the compounds have biological activity. For example, the compounds can be characterized by conventional assays, including but not limited to those assays described below, to determine whether the compounds have a predicted activity, binding activity and/or binding specificity.
Furthermore, high-throughput screening can be used to speed up analysis using such assays. As a result, it can be possible to screen rapidly the molecules described herein for activity, for example, as anti-cancer, anti-bacterial, anti-fungal, anti-parasitic or anti- viral agents. Also, it can be possible to assay how the compounds interact with a ribosome or ribosomal subunit and/or are effective as modulators (for example, inhibitors) of protein synthesis using techniques known in the art. General methodologies for performing high-throughput screening are described, for example, in Devlin, High Throughput Screening, (Marcel Dekker, 1998); and U.S. Patent No. 5,763,263. High-throughput assays can use one or more different assay techniques including, but not limited to, those described below.
(1) Surface Binding Studies. A variety of binding assays can be useful in screening new molecules for their binding activity. One approach includes surface plasmon resonance (SPR) that can be used to evaluate the binding properties of molecules of interest with respect to a ribosome, ribosomal subunit or a fragment thereof.
SPR methodologies measure the interaction between two or more macromolecules in real-time through the generation of a quantum-mechanical surface plasmon. One device, (BIAcore Biosensor RTM from Pharmacia Biosensor, Piscatawy, NJ.) provides a focused beam of polychromatic light to the interface between a gold film (provided as a disposable biosensor "chip") and a buffer compartment that can be regulated by the user. A 100 nm thick "hydro gel" composed of carboxylated dextran that provides a matrix for the covalent immobilization of analytes of interest is attached to the gold film. When the focused light interacts with the free electron cloud of the gold film, plasmon resonance is enhanced. The resulting reflected light is spectrally depleted in wavelengths that optimally evolved the resonance. By separating the reflected polychromatic light into its component wavelengths (by means of a prism), and determining the frequencies that are depleted, the BIAcore establishes an optical interface which accurately reports the behavior of the generated surface plasmon resonance. When designed as
above, the plasmon resonance (and thus the depletion spectrum) is sensitive to mass in the evanescent field (which corresponds roughly to the thickness of the hydrogel). If one component of an interacting pair is immobilized to the hydrogel, and the interacting partner is provided through the buffer compartment, the interaction between the two components can be measured in real time based on the accumulation of mass in the evanescent field and its corresponding effects of the plasmon resonance as measured by the depletion spectrum. This system permits rapid and sensitive real-time measurement of the molecular interactions without the need to label either component.
(2) Fluorescence Polarization. Fluorescence polarization (FP) is a measurement technique that can readily be applied to protein-protein, protein-ligand, or RNA-ligand interactions in order to derive IC50S and Kds of the association reaction between two molecules. In this technique one of the molecules of interest is conjugated with a fluorophore. This is generally the smaller molecule in the system (in this case, the compound of interest). The sample mixture, containing both the ligand-probe conjugate and the ribosome, ribosomal subunit or fragment thereof, is excited with vertically polarized light. Light is absorbed by the probe fluorophores, and re-emitted a short time later. The degree of polarization of the emitted light is measured. Polarization of the emitted light is dependent on several factors, but most importantly on viscosity of the solution and on the apparent molecular weight of the fluorophore. With proper controls, changes in the degree of polarization of the emitted light depends only on changes in the apparent molecular weight of the fluorophore, which in-turn depends on whether the probe-ligand conjugate is free in solution, or is bound to a receptor. Binding assays based on FP have a number of important advantages, including the measurement of IC50S and Kds under true homogenous equilibrium conditions, speed of analysis and amenity to automation, and ability to screen in cloudy suspensions and colored solutions. (3) Protein Synthesis. It is contemplated that, in addition to characterization by the foregoing biochemical assays, the compound of interest can also be characterized as a modulator (for example, an inhibitor of protein synthesis) of the functional activity of the ribosome or ribosomal subunit.
Furthermore, more specific protein synthesis inhibition assays can be performed by administering the compound to a whole organism, tissue, organ, organelle, cell, a cellular or subcellular extract, or a purified ribosome preparation and observing its pharmacological and inhibitory properties by determining, for example, its inhibition constant (IC50) for inhibiting
protein synthesis. Incorporation of 3H leucine or 35S methionine, or similar experiments can be performed to investigate protein synthesis activity. A change in the amount or the rate of protein synthesis in the cell in the presence of a molecule of interest indicates that the molecule is a modulator of protein synthesis. A decrease in the rate or the amount of protein synthesis indicates that the molecule is a inhibitor of protein synthesis.
Furthermore, the compounds can be assayed for anti-proliferative or anti-infective properties on a cellular level. For example, where the target organism is a microorganism, the activity of compounds of interest can be assayed by growing the microorganisms of interest in media either containing or lacking the compound. Growth inhibition can be indicative that the molecule could be acting as a protein synthesis inhibitor. More specifically, the activity of the compounds of interest against bacterial pathogens can be demonstrated by the ability of the compound to inhibit growth of defined strains of human pathogens. For this purpose, a panel of bacterial strains can be assembled to include a variety of target pathogenic species, some containing resistance mecham'sms that have been characterized. Use of such a panel of organisms permits the determination of structure-activity relationships not only in regards to potency and spectrum, but also with a view to obviating resistance mechanisms. The assays can be performed in microtiter trays according to conventional methodologies as published by The National Committee for Clinical Laboratory Standards (NCCLS) guidelines (NCCLS. M7-A5- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Fifth Edition. NCCLS Document M100-S12/M7 (ISBN 1-56238-394-9)).
4. Formulation and Administration
The compounds of the invention can be useful in the prevention or treatment of a variety of human or other animal disorders, including for example, bacterial infection, fungal infections, viral infections, parasitic diseases, and cancer. It is contemplated that, once identified, the active molecules of the invention can be incorporated into any suitable carrier prior to use. The dose of active molecule, mode of administration and use of suitable carrier will depend upon the intended recipient and target organism. The formulations, both for veterinary and for human medical use, of compounds according to the present invention typically include such compounds in association with a pharmaceutically acceptable carrier. The carrier(s) should be "acceptable" in the sense of being compatible with the other ingredients of the formulations and not deleterious to the recipient. Pharmaceutically acceptable carriers, in this regard, are intended to include any and all solvents, dispersion media, coatings,
anti-bacterial and anti-fungal agents, isotonic and absorption delaying agents, and the like, compatible with pharmaceutical administration. The use of such media and agents for pharmaceutically active substances is known in the art. Except insofar as any conventional media or agent is incompatible with the active compound, use thereof in the compositions is contemplated. Supplementary active compounds (identified or designed according to the invention and/or known in the art) also can be incorporated into the compositions. The formulations can conveniently be presented in dosage unit form and can be prepared by any of the methods well known in the art of pharmacy/microbiology. In general, some formulations are prepared by bringing the compound into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
A pharmaceutical composition of the invention should be formulated to be compatible with its intended route of administration. Examples of routes of administration include oral or parenteral, for example, intravenous, intradermal, inhalation, transdermal (topical), transmucosal, and rectal administration. Solutions or suspensions used for parenteral, intradermal, or subcutaneous application can include the following components: a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerine, propylene glycol or other synthetic solvents; antibacterial agents such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid; buffers such as acetates, citrates or phosphates and agents for the adjustment of tonicity such as sodium chloride or dextrose. pH can be adjusted with acids or bases, such as hydrochloric acid or sodium hydroxide.
Useful solutions for oral or parenteral administration can be prepared by any of the methods well known in the pharmaceutical art, described, for example, in Remington's Pharmaceutical Sciences, 18th ed. (Mack Publishing Company, 1990). Formulations for parenteral administration can also include glycocholate for buccal administration, methoxysalicylate for rectal administration, or citric acid for vaginal administration. The parenteral preparation can be enclosed in ampoules, disposable syringes or multiple dose vials made of glass or plastic. Suppositories for rectal administration also can be prepared by mixing the drug with a non-irritating excipient such as cocoa butter, other glycerides, or other compositions which are solid at room temperature and liquid at body temperatures.
Formulations also can include, for example, polyalkylene glycols such as polyethylene glycol, oils of vegetable origin, and hydrogenated naphthalenes. Formulations for direct administration
can include glycerol and other compositions of high viscosity. Other potentially useful parenteral carriers for these drugs include ethylene-vinyl acetate copolymer particles, osmotic pumps, implantable infusion systems, and liposomes. Formulations for inhalation administration can contain as excipients, for example, lactose, or can be aqueous solutions containing, for example, polyoxyethylene-9-lauryl ether, glycocholate and deoxycholate, or oily solutions for administration in the form of nasal drops, or as a gel to be applied intranasally. Retention enemas also can be used for rectal delivery.
Formulations of the present invention suitable for oral administration can be in the form of: discrete units such as capsules, gelatin capsules, sachets, tablets, troches, or lozenges, each containing a predetermined amount of the drug; a powder or granular composition; a solution or a suspension in an aqueous liquid or non-aqueous liquid; or an oil-in- water emulsion or a water- in-oil emulsion. The drug can also be administered in the form of a bolus, electuary or paste. A tablet can be made by compressing or molding the drag optionally with one or more accessory ingredients. Compressed tablets can be prepared by compressing, in a suitable machine, the drag in a free-flowing form such as a powder or granules, optionally mixed by a binder, lubricant, inert diluent, surface active or dispersing agent. Molded tablets can be made by molding, in a suitable machine, a mixture of the powdered drag and suitable carrier moistened with an inert liquid diluent.
Oral compositions generally include an inert diluent or an edible carrier. For the purpose of oral therapeutic administration, the active compound can be incorporated with excipients. Oral compositions prepared using a fluid carrier for use as a mouthwash include the compound in the fluid carrier and are applied orally and swished and expectorated or swallowed. Pharmaceutically compatible binding agents, and/or adjuvant materials can be included as part of the composition. The tablets, pills, capsules, troches and the like can contain any of the following ingredients, or compounds of a similar nature: a binder such as microcrystalline cellulose, gum tragacanth or gelatin; an excipient such as starch or lactose; a disintegrating agent such as alginic acid, Primogel, or corn starch; a lubricant such as magnesium stearate or Sterotes; a glidant such as colloidal silicon dioxide; a sweetening agent such as sucrose or saccharin; or a flavoring agent such as peppermint, methyl salicylate, or orange flavoring. Pharmaceutical compositions suitable for injectable use include sterile aqueous solutions
(where water soluble) or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersion. For intravenous administration, suitable carriers
include physiological saline, bacteriostatic water, Cremophor ELTM (BASF, Parsippany, NJ) or phosphate buffered saline (PBS). It should be stable under the conditions of manufacture and storage and should be preserved against the contaminating action of microorganisms such as bacteria and fungi. The carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyetheylene glycol), and suitable mixtures thereof. The proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants, hi many cases, it will be preferable to include isotonic agents, for example, sugars, polyalcohols such as manitol, sorbitol, sodium chloride in the composition. Prolonged absorption of the injectable compositions can be brought about by including in the composition an agent which delays absorption, for example, aluminum monostearate and gelatin.
Sterile injectable solutions can be prepared by incorporating the active compound in the required amount in an appropriate solvent with one or a combination of ingredients enumerated above, as required, followed by filter sterilization. Generally, dispersions are prepared by incorporating the active compound into a sterile vehicle which contains a basic dispersion medium and the required other ingredients from those enumerated above, hi the case of sterile powders for the preparation of sterile injectable solutions, methods of preparation include vacuum drying and freeze-drying which yields a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
Formulations suitable for intra-articular administration can be in the form of a sterile aqueous preparation of the drug that can be in microcrystalline form, for example, in the form of an aqueous microcrystalline suspension. Liposomal formulations or biodegradable polymer systems can also be used to present the drug for both intra-articular and ophthalmic administration.
Formulations suitable for topical administration, including eye treatment, include liquid or semi-liquid preparations such as liniments, lotions, gels, applicants, oil-in-water or water-in- oil emulsions such as creams, ointments or pastes; or solutions or suspensions such as drops. Formulations for topical administration to the skin surface can be prepared by dispersing the drug with a dermatologically acceptable carrier such as a lotion, cream, ointment or soap. Particularly useful are carriers capable of forming a film or layer over the skin to localize application and inhibit removal. For topical administration to internal tissue surfaces, the agent
can be dispersed in a liquid tissue adhesive or other substance known to enhance adsorption to a tissue surface. For example, hydroxypropylcellulose or fibrinogen/thrombin solutions can be used to advantage. Alternatively, tissue-coating solutions, such as pectin-containing formulations can be used. For inhalation treatments, inhalation of powder (self-propelling or spray formulations) dispensed with a spray can, a nebulizer, or an atomizer can be used. Such formulations can be in the form of a fine powder for pulmonary administration from a powder inhalation device or self- propelling powder-dispensing formulations. In the case of self-propelling solution and spray formulations, the effect can be achieved either by choice of a valve having the desired spray characteristics (i.e., being capable of producing a spray having the desired particle size) or by incorporating the active ingredient as a suspended powder in controlled particle size. For administration by inhalation, the compounds also can be delivered in the form of an aerosol spray from pressured container or dispenser which contains a suitable propellant, e.g., a gas such as carbon dioxide, or a nebulizer. Systemic administration also can be by transmucosal or transdermal means. For transmucosal or transdermal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants generally are known in the art, and include, for example, for transmucosal administration, detergents and bile salts. Transmucosal administration can be accomplished through the use of nasal sprays or suppositories. For transdermal administration, the active compounds typically are formulated into ointments, salves, gels, or creams as generally known in the art.
The active compounds can be prepared with carriers that will protect the compound against rapid elimination from the body, such as a controlled release formulation, including implants and microencapsulated delivery systems. Biodegradable, biocompatible polymers can be used, such as ethylene vinyl acetate, polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic acid. Methods for preparation of such formulations will be apparent to those skilled in the art. Liposomal suspensions can also be used as pharmaceutically acceptable carriers. These can be prepared according to methods known to those skilled in the art, for example, as described in U.S. Patent No. 4,522,811. Oral or parenteral compositions can be formulated in dosage unit form for ease of administration and uniformity of dosage. Dosage unit form refers to physically discrete units suited as unitary dosages for the subject to be treated; each unit containing a predetermined
quantity of active compound calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier. The specification for the dosage unit forms of the invention are dictated by and directly dependent on the unique characteristics of the active compound and the particular therapeutic effect to be achieved, and the limitations inherent in the art of compounding such an active compound for the treatment of individuals. Furthermore, administration can be by periodic injections of a bolus, or can be made more continuous by intravenous, intramuscular or intraperitoneal administration from an external reservoir (e.g., an intravenous bag).
Where adhesion to a tissue surface is desired the composition can include the drug dispersed in a fibrinogen-thrombin composition or other bioadhesive. The compound then can be painted, sprayed or otherwise applied to the desired tissue surface. Alternatively, the drugs can be formulated for parenteral or oral administration to humans or other mammals, for example, in effective amounts, e.g., amounts that provide appropriate concentrations of the drug to target tissue for a time sufficient to induce the desired effect. Where the active compound is to be used as part of a transplant procedure, it can be provided to the living tissue or organ to be transplanted prior to removal of tissue or organ from the donor. The compound can be provided to the donor host. Alternatively or, in addition, once removed from the donor, the organ or living tissue can be placed in a preservation solution containing the active compound. In all cases, the active compound can be administered directly to the desired tissue, as by injection to the tissue, or it can be provided systemically, either by oral or parenteral administration, using any of the methods and formulations described herein and/or known in the art. Where the drug comprises part of a tissue or organ preservation solution, any commercially available preservation solution can be used to advantage. For example, useful solutions known in the art include Collins solution, Wisconsin solution, Belzer solution, Eurocollins solution and lactated Ringer's solution.
Active compound as identified or designed by the methods described herein can be administered to individuals to treat disorders (prophylactically or therapeutically), hi conjunction with such treatment, pharmacogenomics (i.e., the study of the relationship between an individual's genotype and that individual's response to a foreign compound or drug) can be considered. Differences in metabolism of therapeutics can lead to severe toxicity or therapeutic failure by altering the relation between dose and blood concentration of the pharmacologically active drug. Thus, a physician or clinician can consider applying knowledge obtained in relevant
pliarmacogenomics studies in deteraiining whetlier to administer a drag as well as tailoring the dosage and/or therapeutic regimen of treatment with the drug.
In therapeutic use for treating, or combating, bacterial infections in mammals, the compounds or pharmaceutical compositions thereof will be administered orally, parenterally and/or topically at a dosage to obtain and maintain a concentration, that is, an amount, or blood- level or tissue level of active component in the animal undergoing treatment which will be anti- microbially effective. Generally, an effective amount of dosage of active component will be in the range of from about 0.1 to about 100, more preferably from about 1.0 to about 50 mg/kg of body weight/day. The amount administered will also likely depend on such variables as the type and extent of disease or indication to be treated, the overall health status of the particular patient, the relative biological efficacy of the compound delivered, the formulation of the drug, the presence and types of excipients in the formulation, and the route of administration. Also, it is to be understood that the initial dosage administered can be increased beyond the above upper level in order to rapidly achieve the desired blood-level or tissue level, or the initial dosage can be smaller than the optimum and the daily dosage can be progressively increased during the course of treatment depending on the particular situation. If desired, the daily dose can also be divided into multiple doses for administration, for example, two to four times per day.
5. Examples
In the following examples, nuclear magnetic resonance (NMR) spectra were obtained on a Bruker Avance 300 or Avance 500 spectrometer, or in some cases a GE-Nicolet 300 spectrometer. Common reaction solvents were either high performance liquid chromatography (HPLC) grade or American Chemical Society (ACS) grade, and anhydrous as obtained from the manufacturer unless otherwise noted. "Chromatography" or "purified by silica gel" refers to flash column chromatography using silica gel (EM Merck, Silica Gel 60, 230-400 mesh) unless otherwise noted.
Exemplary compounds synthesized in accordance with the invention are listed in Table 1. A straight, unbolded or unhatched bond from a chiral center indicates that the substituent can be either an enantiomer, i.e., R or S, or a mixture of both. A wavy bond indicates that the substituent can be either cis or trans, or a mixture of both. Furthermore, the chemical names are provided for convenience and are not intended to limit the indicated chemical structures. To the extent that there is a discrepancy between the chemical name and the structure of a compound, the structure of the compound shall govern. Also, depending on the conventions and choices
available, more than one chemical name can be given to a particular chemical structure. As a nonlimiting example, Compound 101, below, is drawn with stereochemistry indicated for the methyl acetamide substituent on the oxazolidinone ring. Compound 101 is named indicating the "5S" stereochemistry at the chiral carbon center at which the acetamide substituent is attached.
The compounds of the present invention can be prepared, formulated, and delivered as salts, esters, and prodrugs. For convenience, the compounds in Table 1 are generally shown without indicating a particular salt, ester, or prodrug form and are generally named in the table without further limitation to such salts, esters, or prodrugs. As a nonlimiting example, Compound 133, having a tertiary amine group, is depicted as the free base compound, although it can be prepared as the hydrochloride salt, as indicated from the synthetic procedure provided for the compound in Example 28.
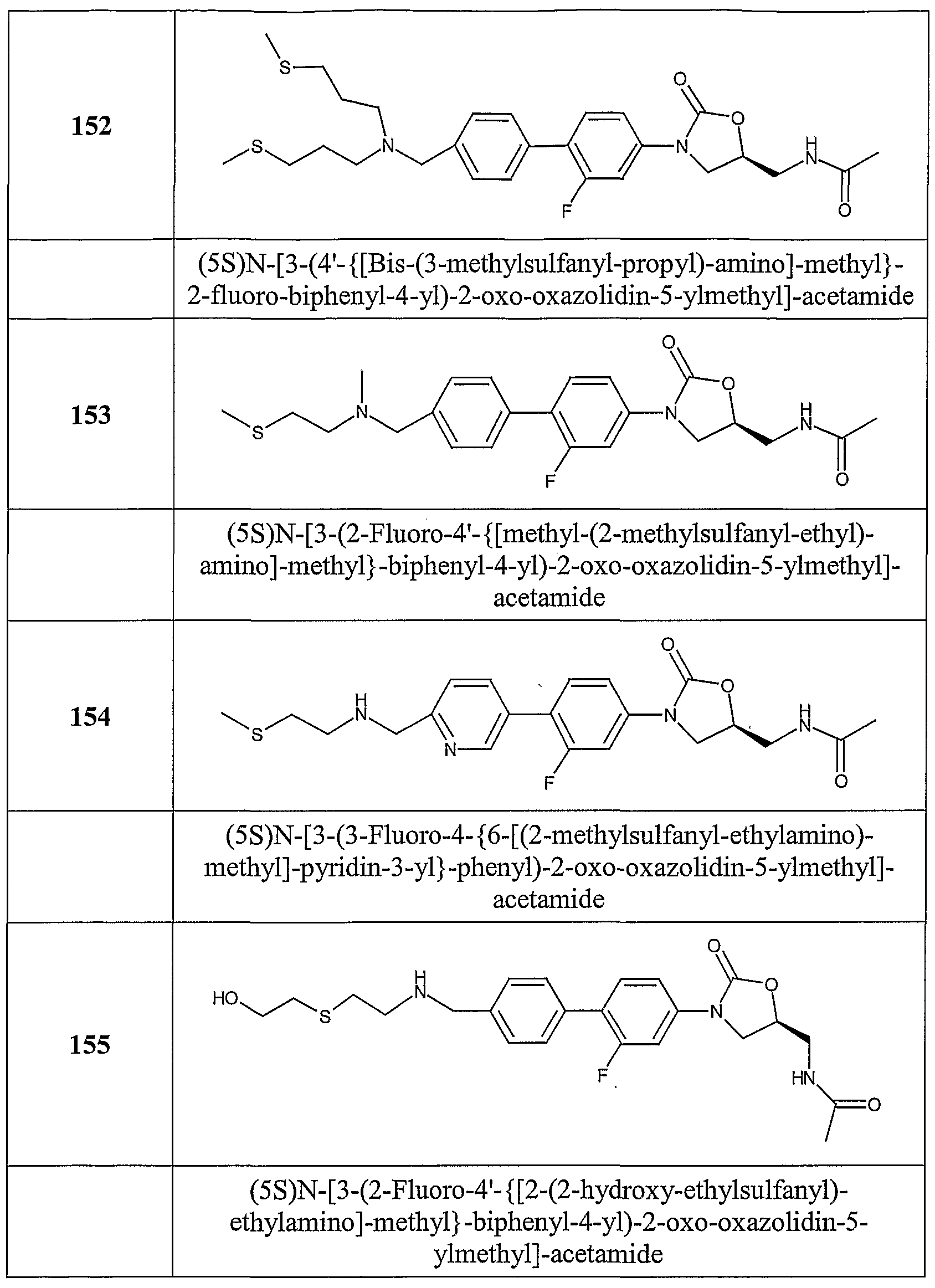
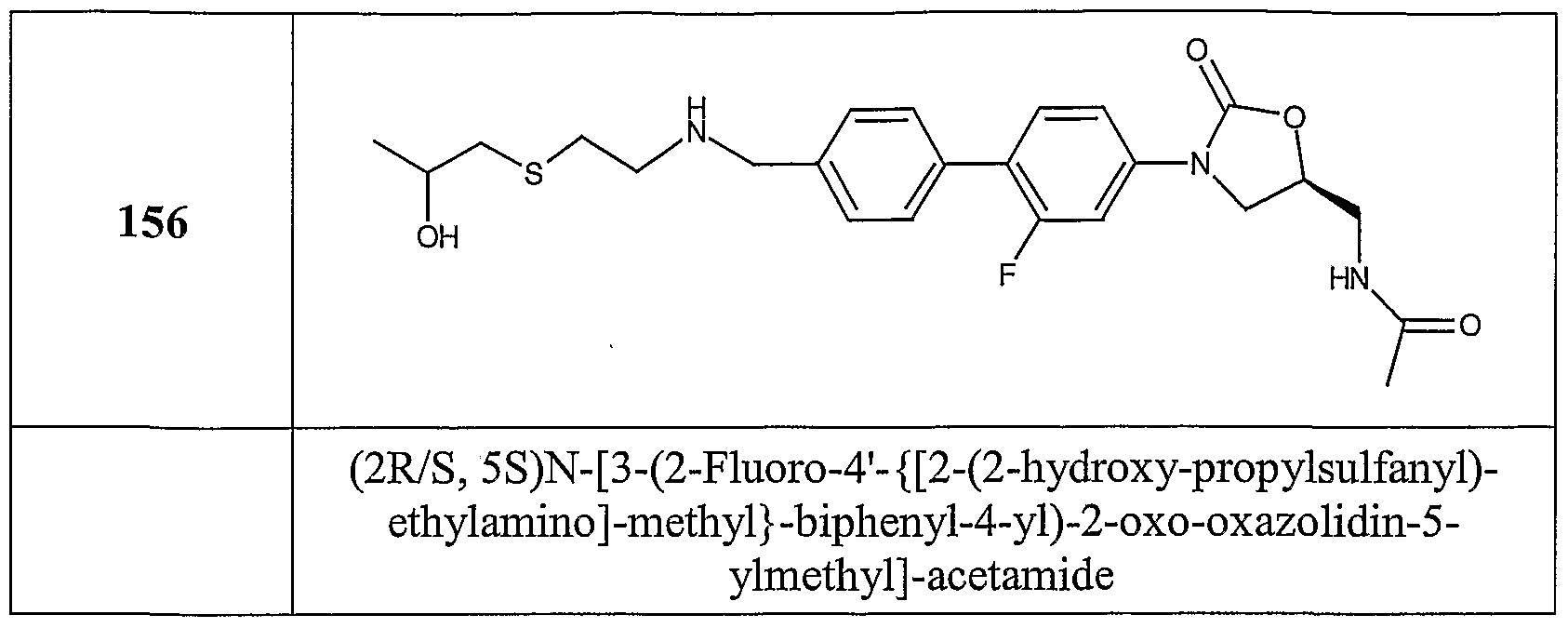
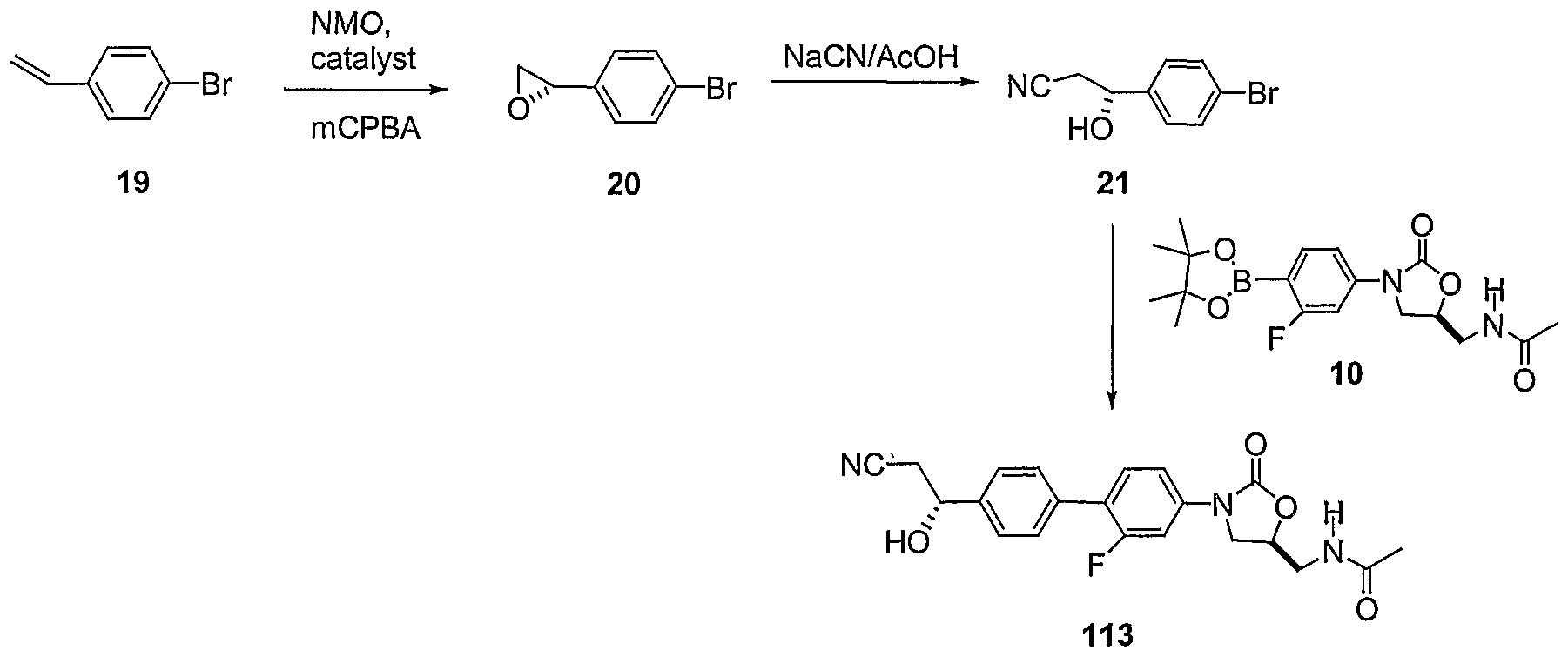
Table 1
The following schemes depict some exemplary chemistries available for synthesizing the compounds of the invention. It will be appreciated, however, that the desired compounds may be synthesized using other alternative chemistries known and used in the art.
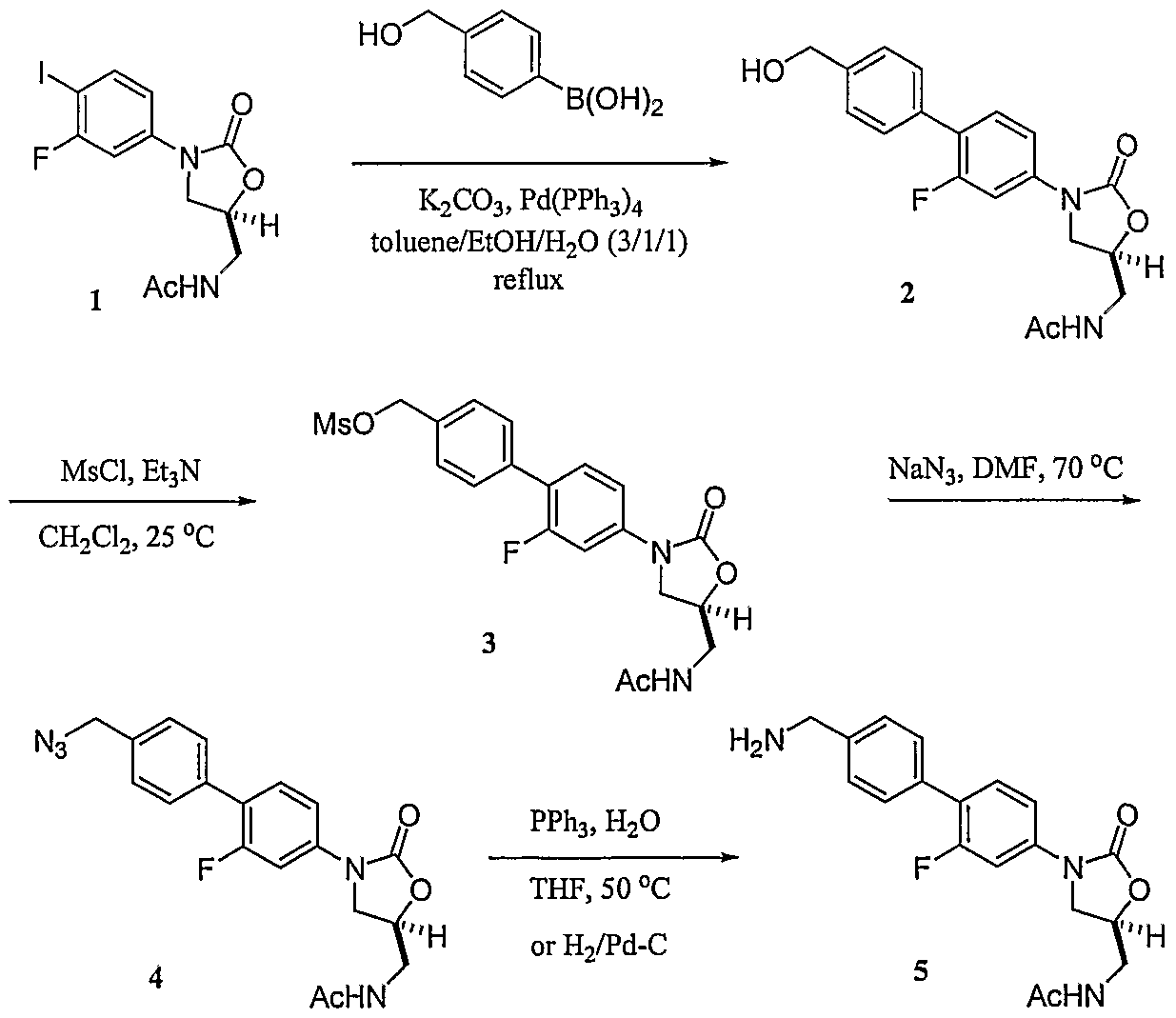
Example 1 - Synthesis of Biaryl Precursors
Scheme 1 depicts the synthesis of various biaryl intermediates useful in producing compounds of the present invention. Known iodoaryl oxazolidinone intermediate 1 {see U.S. Patent Nos. 5,523,403 and 5,565,571) is coupled to a substituted aryl boronic acid (the Suzuki reaction) to produce biaryl alcohol 2. Mesylate 3, azide 4, and amine 5 are then synthesized using chemistries well known to those skilled in the art.
Scheme 1
A suspension of N-[3-(3-fluoro-4-iodo-phenyl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (oxazolidinone intermediate 1, 14.0 g, 37 mmol) in toluene (120 mL) was treated with 4- (hydroxymethyl) phenylboronic acid (7.87 g, 51.8 mmol, 1.4 equiv), potassium carbonate (K2CO3, 15.32 g, 111 mmol, 3.0 equiv), etlianol (EtOH, 40 mL), and water (H2O, 40 mL) at 25 0C, and the resulting mixture was degassed three times under a steady stream of argon at 25 0C. Tetrakis(triphenylρhosphine)palladium (Pd(PPh3)4, 2.14 g, 1.85 mmol, 0.05 equiv) was subsequently added to the reaction mixture, and the resulting reaction mixture was degassed three times again before being warmed to gentle reflux for 6 hours (hr). When thin layer chromatography (TLC) and HPLC showed the coupling reaction was complete, the reaction mixture was cooled to room temperature before being treated with H2O (240 mL). The resulting mixture was then stirred at room temperature for 10 minutes (min) before being cooled to 0-5 0C for 1 hr. The solid precipitates were collected by filtration, washed with H2O (2 x 100 mL) and 20% ethyl acetate (EtOAc)/hexane (2 x 50 mL), and dried in vacuo. The crude desired iV-[3-(2- Fluoro-4'-hydroxymethyl-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (alcohol 2, 12.50 g, 94% yield) was obtained as off-white solids. This material was found to be essentially pure by HPLC and 1H NMR and was directly used in the subsequent reaction without further purification. 1H NMR (300 MHz, DMSO-^6) δ 1.76 (s, 3H, COCH3), 3.35 (t, 2Η, J= 5.4 Hz), 3.69 (dd, IH, J= 6.4, 9.2 Hz), 4.08 (t, IH, J= 9.1 Hz), 4.46 (d, 2H, J= 5.7 Hz, CH2OH), 4.68 (m, IH), 5.16 (t, IH, J- 5.7 Hz, OH), 7.25 - 7.52 (m, 7Η, aromatic-H), 8.18 (t, 1Η, J= 5.8 Hz, NHCOCH3). LCMS (ESI) mle 359 (M + H)+.
Synthesis of mesylate 3
A suspension of alcohol 2 (12.49 g, 34.90 mmol) in methylene chloride (CH2Cl2, 150 mL) was treated with triethylamine (Et3N, 7.07 g, 9.7 mL, 70 mmol, 2.0 equiv) at 25 0C, and the resulting mixture was cooled to 0-5 0C before being treated dropwise with methanesulfonyl chloride (MsCl, 4.80 g, 3.24 mL, 41.9 mmol, 1.2 equiv) at 0-5 0C. The resulting reaction mixture was subsequently stirred at 0-5 0C for 2 hr. When TLC and HPLC showed the reaction was complete, the reaction mixture was treated with H2O (100 mL) at 0-5 0C. The mixture was then concentrated in vacuo to remove most of the CH2Cl2, and the resulting slurry was treated with H2O (150 mL). The mixture was stirred at room temperature for 10 min before being cooled to 0-5 0C for 30 min. The solid precipitates were collected by filtration, washed with H2O (2 x 100 mL) and 20% EtOAc/hexane (2 x 50 mL), and dried in vacuo. The crude desired
methanesulfonic acid 4'-[5-(acetylamino-methyl)-2-oxo-oxazolid.in-3-yl]-2l-fluoro-biphenyl-4- ylmethyl ester (mesylate 3, 11.84 g, 78% yield) was obtained as off-white solids, which by TLC and HPLC was found to be essentially pure and was directly used in the subsequent reaction without further purification. LCMS (ESI) mle A2>1 (M + H)+. Synthesis of azide 4
A solution of mesylate 3 (9.27 g, 21.26 mmol) in anhydrous N,N-dimethylformamide (DMF, 50 mL) was treated with sodium azide (NaN3, 5.53 g, 85.04 mmol, 4.0 equiv) at 25 0C, and the resulting reaction mixture was warmed to 70-80 0C for 4 hr. When TLC and HPLC showed the reaction was complete, the reaction mixture was cooled to room temperature before being treated with H2O (150 mL). The resulting mixture was stirred at room temperature for
10 min before being cooled to 0-5 0C for 1 hr. The solid precipitates were collected by filtration, washed with H2O (2 x 100 mL) and 20% EtOAc/hexane (2 x 50 mL), and dried in vacuo. The crude desired N-[3-(4'-azidomethyl-2-fluoro-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]- acetamide (azide 4, 7.16 g, 88 % yield) was obtained as off-white solids. The material was found to be essentially pure by TLC and HPLC and was directly used in the subsequent reaction without further purification. LCMS (ESI) mle 384 (M + H)+.
Synthesis of amine 5
A solution of azide 4 (7.16 g, 18.69 mmol) in tetrahydrofuran (THF, 100 mL) was treated with triphenylphosphine (PPh3, 5.88 g, 22.43 mmol, 1.2 equiv) and H2O (3.6 g, 3.6 mL, 0.2 mmol, 11.0 equiv) at 25 0C, and the resulting reaction mixture was warmed to 50-55 0C for 12 hr. When TLC and HPLC showed the reduction reaction was complete, the reaction mixture was cooled to room temperature before the solvents were removed in vacuo. The residue was directly purified by flash column chromatography (0-15% methanol (MeOH)-CH2Cl2 gradient elution) to afford the desired iV-[3-(4'-Aminomethyl-2-fluoro-biphenyl-4-yl)-2-oxo-oxazolidin-5- ylmethylj-acetamide (amine 5, 5.82 g, 87% yield) as off-white crystals, which were of sufficient purity to be directly used in subsequent reactions. 1H ΝMR (300 MHz, DMSO- J6) δ 1.85 (s, 3H, COCH3), 3.04 (br. s, 2H, NH2), 3.44 (t, 2Η, J= 5.4 Hz), 3.78 (m, 3H), 4.18 (t, IH, J= 9.1 Hz), 4.77 (m, IH), 7.25 - 7.60 (m, 7H, aromatic-H), 8.20 (t, 1Η, J= 5.8 Hz, NHCOCH3). LCMS (ESI) mle 359 (M + 2H)2+.
Example 2 - Synthesis of Compound 101
Scheme 2
A suspension of 1.884 g (5.3 mmol) (55)-iV-[3-(2-fluoro-4'-formyl-biphenyl-4-yl)-2-oxo- oxazolidin-5-ylmethyl]-acetamide (aldehyde 6, made from oxazolidinone intermediate 1 and 4- formylboronic acid in the same fashion as the synthesis of alcohol 2 in Example 1) in MeOH (25 mL) and H2O (25 mL) was treated with sodium cyanide (NaCN, 312 mg, 6.36 mmol) and ammonium chloride (NH4Cl, 340 mg, 6.36 mmol) at room temperature. The resulting reaction mixture was stirred at room temperature for 30 min before being warmed to 50 0C for 1 hr. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was cooled to room temperature and treated with H2O (25 mL). The mixture was stirred at room temperature for 30 min before being cooled to 0-5 0C for 1 hr. The solids were collected by filtration, washed with H2O (2 x 25 mL), and dried in vacuo to afford 1.80 g of the crude desired (5iS)-N-{3-[4'-(amino-cyano-methyl)-2-fluoro-biphenyl-4-yl]-2-oxo-oxazolidin-5-yhnethyl}- acetamide (compound 101, 89 % yield) as an off-white powder. The product was found to be essentially pure by HPLC and 1HNMR. LCMS (EI): mle 383 (M+ + H).
Example 3 - Synthesis of Compounds 104 and 105
Scheme 3
104
Ac2O, DMAP, Et3N, DMF
105
The hydrochloride salt of amine 5 (0.40 g, 1.02 mmol) was dissolved in DMF (10 niL) and Hunig's base (diisopropylethylamine, 1 mL). To this solution was added bromoacetonitrile 7 (0.08 mL, 1.11 mmol) and the mixture was stirred for 1 hr at room temperature. The solvent was evaporated and the crude was partitioned between dilute ammonium hydroxide (NH4OH, 30 mL) and 15 % MeOH in CH2Cl2 (30 mL). The aqueous layer was back extracted with 15 % MeOH in CH2Cl2 (2 x 30 mL), the combined organic layer was dried over sodium sulfate (Na2SO4), and the solvent was evaporated. The crude was purified on silica gel (first eluting with 30:1 CH2Cl2MeOH, then with 30:1:0.04 CH2Cl2/MeOH/NH4OH). The isolated solid was triturated with 1:1 diethyl ether (Et2O)/acetonitrile (20 mL) and filtered to give compound 104 as a white crystalline solid (0.237g, 59 % yield). LCMS (ESI): m/e 397.0 (M + H)+.
Compound 104 (0.10 g, 0.23 mmol) and a catalytic amount of 4-dimethylaminopyridme (DMAP) were dissolved in DMF (3 mL) and Et3N (0.3 mL). To this solution was added acetic anhydride (0.05 mL, 0.46 mmol) and the mixture was stirred at room temperature overnight. The solvent was evaporated, the residue was dissolved in CH2Cl2 (20 mL), and the solution was extracted with IN HCl (2 x 20 mL) and H2O (10 mL). The organic layer was dried over Na2SO4 and the solvent evaporated to give compound 105 as a white solid in quantitative yield. LCMS (ESI): m/e 439.1 (M + H)+.
Example 4 - Synthesis of Compound 107 To a suspension of the hydrochloride salt of amine 5 (150 mg, 0.382 mmol), cyanoacetic acid (40 mg, 0.458 mmol) and l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI, 87.8 mg, 0.458 mmol) in DMF (8 mL) was added Hunig's base (0.252 mL, 1.528 mmol) and the mixture was stirred at ambient temperature overnight. The reaction mixture was concentrated, precipitated with ether and the solid thus obtained was washed with 10% aqueous NH4OH. The crude solid thus obtained was purified by flash chromatography (15:1:0.01 CH2Cl2/MeOH/NH4OH) to yield 37 mg of compound 107. LCMS (ESI): m/e 425 (M+H+).
Example 5 - Synthesis of Compound 109
Scheme 4
A solution of 2,5-dibromopyridine 8 (25 g, 105.5 mmol) in toluene (1.24 L) was cooled to -78 0C before being treated dropwise with a 2.5 M solution of n-butyl lithium (n-BuLi) in hexane (50.6 niL, 126.6 mmol) at -78 0C. The resulting reaction mixture was stirred at -78 0C for 1 hr before being treated with anhydrous DMF (11.6 g, 12.2 niL, 158.0 mmol) at -78 0C. The reaction mixture was stirred at -78 0C for an additional 1 hr before being gradually warmed to room temperature for 6 hr. When TLC and HPLC showed that the reaction was complete, the reaction mixture was quenched with H2O (200 mL). The two layers were separated, and the aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic extracts were then washed with H2O (2 x 200 mL) and saturated aqueous NaCl (100 mL) and dried over magnesium sulfate (MgSO4). The solvents were then removed in vacuo, and the residual pale- yellow oil was purified by flash column chromatography (0-15% EtOAc-hexane gradient elution) to afford the desired 5-bromo-pyridine-2-carbaldehyde 9 (10.2 g, 52% yield ) as pale- yellow solids.
A suspension of oxazolidnone intermediate 1 (20.0 g, 52.8 mmol) in anhydrous 1,4- dioxane (130 mL) was treated with 4,4,5, 5-tetramethyl-[l,3,2]dioxaborolane (10.2 g, 11.6 mL, 80.0 mmol) and Et3N (16.0 g, 22.4 mL, 158.4 mmol) at room temperature, and the resulting reaction mixture was degassed three times under a steady stream of argon before being treated
with dichloro[l,r-bis(diphenylphosphino)ferrocene] palladium (II) (Pd(dppf)2Cl2, 1.32 g, 1.6 mmol) at room temperature. The reaction mixture was then degassed three times again under a steady stream of argon before being heated to reflux for 7 hr. When TLC and LCMS showed that the reaction was complete, the reaction mixture was cooled to room temperature before being treated with H2O (100 mL) and EtOAc (100 mL). The two layers were separated, and the aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic extracts were washed with H2O (2 x 50 mL) and saturated aqueous NaCl (50 mL), dried over MgSO4, and concentrated in vacuo. The residual brown oil was further dried in vacuo to afford the crude desired N-{3-[3-fluoro-4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-phenyl]-2-oxo- oxazolidin-5-ylmethyl}acetamide (boronate 10, 18.8 g, 94%) as a brown solid which was of sufficient purity to be used in subsequent reactions.
A solution of boronate 10 (11.05 g, 29.2 mmol) and 5-bromo-pyridine-2-carbaldehyde 9 (4.185 g, 22.5 mmol) in toluene (150 mL) was treated with solid K2CO3 (9.315 g, 67.5 mmol), EtOH (50 mL) and H2O (50 mL) at room temperature. The resulting reaction mixture was degassed three times under argon before being treated with Pd(dppf)2Cl2 (564 mg, 0.675 mmol) at room temperature. The reaction mixture was then degassed three times again under argon before being warmed to reflux for 2 hr. When HPLC showed that the reaction was complete, the reaction mixture was cooled to room temperature before being treated with H2O (200 mL) and EtOAc (100 mL). The two layers were separated, and the aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic extracts were washed with H2O (2 x 50 mL) and saturated aqueous NaCl (50 mL), dried over MgSO4, and concentrated in vacuo. The residue was then purified by flash column chromatography (0-5% MeOH-CH2Cl2 gradient elution) to afford the desired (5S)-N- {3-[3-fluoro-4-(6-formyl-pyridin-3-yl)-phenyl]-2-oxo-oxazolidin-5- ylmethyl}-acetamide (aldehyde 11, 4.42 g, 55% yield) as gray solids. A solution of aldehyde 11 (180 mg, 0.5 mmol) in THF (5 mL) and DMF (3 mL) was treated with aminoacetonitrile bissulfate (93 mg, 0.6 mmol) and sodium triacetoxyborohydride (NaB(OAc)3H, 212 mg, 1.0 mmol) at 25 0C, and the resulting reaction mixture was stirred at 25 0C for an additional 4 hr. When TLC and HPLC showed that the reaction was complete, the reaction mixture was treated with H2O (5 mL) and EtOAc (20 mL). The two layers were separated, and the aqueous layer was extracted with EtOAc (2 x 10 mL). The combined organic extracts were washed with H2O (5 mL) and saturated aqueous NaCl (5 mL), dried over MgSO4, and concentrated in vacuo. The residue was then purified by flash column chromatography
(0-5% MeOH-CH2Cl2 gradient elution) to afford the desired (5»S)-N-[3-(4-{6-[(cyanomethyl- amino)-methyl]-pyridin-3-yl}-3-fluoro-phenyl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (compound 109, 169 mg, 85%) as white solids. LCMS (EI): mle 398 (M+ + H).
Example 6 - Synthesis of Compound 112
Scheme 5
12 13 14
A solution of D-p-hydroxyphenylglycine 12 (23.8 g, 142.3 mmol) and K
2CO
3 (39.3 g, 0.070 niL, 284.6 mmol) in THF (200 niL) and H
2O (200 niL) was treated with di-tert-butyl dicarbonate (BOC
2O, 34.14 g, 156.6 mmol) at 25
0C and the resulting reaction mixture was stirred at 25
0C for 2 hr. When TLC and HPLC showed the reaction was complete, the reaction mixture was treated with EtOAc (200 mL) and H
2O (200 mL). The two layers were separated, and the aqueous solution was extracted with EtOAc (200 mL). The aqueous layer was then acidified with a 2 N HCl aqueous solution to pH 4 before being extracted with EtOAc (2 x 200 mL). The combined organic extracts were then washed with H
2O (2 x 100 mL) and saturated aqueous NaCl (100 mL), dried over MgSO
4, and concentrated in vacuo. The residual white solids were further dried in vacuo to afford crude desired tert-butoxycarbonylamino-(4-hydroxy- phenyl)-acetic acid (Boc-protected intermediate 13, 36.5 g, 96% yield), which was found to be essentially pure and was directly used in subsequent reactions without further purification.
A solution of Boc-protected intermediate 13 (4.005 g, 15 mmol) in anhydrous THF (20 mL) was treated dropwise with a 1 M solution OfBH3-THF in THF (30 mL, 30 mmol) at 0-5 0C, and the resulting reaction mixture was stirred at 0-5 0C for an additional 2 hr. When TLC and HPLC showed that the reduction reaction was complete, the reaction mixture was treated with H2O (50 mL) and EtOAc (50 mL). The mixture was then stirred at 25 0C for 30 min before being separated, and the aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic extracts were then washed with H2O (2 x 20 mL) and saturated aqueous NaCl (20 mL), dried over MgSO4, and concentrated in vacuo. The residue was then directly purified by flash column chromatography (0-5 % MeOH-CH2Cl2 gradient elution) to afford the desired [2- hydroxy-l-(4-hydroxy-phenyl)-ethyl]-carbamic acid tert-butyl ester (alcohol 14, 2.50 g, 66% yield) as a white powder which was directly used in subsequent reactions without further purification. A suspension of alcohol 14 (670 mg, 2.65 mmol) in CH2Cl2 (10 mL) was treated with N- phenyltrifluoromethane sulfonamide (947 mg, 2.65 mmol) and Et3N (535.3 mg, 0.74 mL, 5.3 mmol) at 25 0C, and the resulting reaction mixture was stirred at 25 0C for an additional 2 hr. When TLC and HPLC showed that the reduction reaction was complete, the reaction mixture was quenched with H2O (10 mL) and the mixture was stirred at room temperature for 30 min. The two layers were then separated, and the aqueous layer was extracted with CH2Cl2 (2 x 20 mL). The combined organic extracts were then washed with H2O (2 x 10 mL) and saturated aqueous NaCl (10 mL), dried over MgSO4, and concentrated in vacuo. The residue was then
directly purified by flash column chromatography (0-5 % MeOH-CH2Cl2 gradient elution) to afford the desired trifluoromethanesulfonic acid 4-(l-tert-butoxycarbonylamino-2- hydroxyethyl)phenyl ester (intermediate 15, 945 mg, 92% yield) as a white powder which was directly used in subsequent reactions without further purification. A solution of boronate 10 (2.162 g, 5.72 mmol) and intermediate 15 (1.70 g, 4.4 mmol) in toluene (24 mL) was treated with solid K2CO3 (1.82 g, 13.2 mmol), EtOH (8.0 mL) and H2O (8.0 mL) at room temperature. The resulting reaction mixture was degassed three times under argon before being treated with Pd(dppf)2Cl2 (184 mg, 0.22 mmol) at room temperature. The reaction mixture was then degassed three times again under argon before being warmed to reflux for 2 hr. When HPLC showed that the reaction was complete, the reaction mixture was cooled to room temperature before being treated with H2O (20 mL) and EtOAc (20 mL). The two layers were separated, and the aqueous layer was extracted with EtOAc (2 x 20 mL). The combined organic extracts were washed with H2O (2 x 20 mL) and saturated aqueous NaCl (20 mL), dried over MgSO4, and concentrated in vacuo. The residue was then purified by flash column chromatography (0-5 % MeOH-CH2Cl2 gradient elution) to afford the' desired (1 - {4'-[5-
(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2'-fluoro-biphenyl-4-yl}-2-hydroxyethyl)carbamic acid ester (biaryl intermediate 16, 1.543 g, 72% yield) as a yellow oil, which solidified upon standing at room temperature in vacuo.
A suspension of biaryl intermediate 16 (694 mg, 1.425 mmol) in anhydrous CH2Cl2 (10 mL) was treated with Hunig's base (388 mg, 0.522 mL, 2.90 mmol) and MsCl (196 mg, 0.132 mL, 1.71 mmol) at 0-5 0C, and the resulting reaction mixture was stirred at 0-5 0C for an additional 2 hr. When TLC and HPLC showed that the reaction was complete, the reaction mixture was quenched with H2O (10 mL). The two layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 x 10 mL). The combined organic extracts were washed with H2O (2 x 10 mL) and saturated aqueous NaCl (10 mL), dried over MgSO4, and concentrated in vacuo. The residue was then purified by flash column chromatography (0-5 % MeOH-CH2Cl2 gradient elution) to afford the desired methanesulfonic acid 2-{4'-[5-(acetylamino-methyl)-2-oxo- oxazolidin-3-yl]-2'-fluoro-biphenyl-4-yl}-2-tert-butoxycarbonylamino-ethyl ester (mesylate 17, 647 mg, 80.3% yield) as pale-yellow solids, which was directly used in the subsequent reactions. A solution of mesylate 17 (283 mg, 0.5 mmol) in methyl sulfoxide (DMSO, 4.0 mL) was treated with sodium cyanide (NaCN, 74 mg, 1.5 mmol), 18-crown-6 (10 mg, cat.), and potassium iodide (KI, 10 mg) at room temperature. The resulting reaction mixture was then warmed to 90-100 0C for 24 hr. When TLC and HPLC showed that the reaction was almost complete, the
reaction mixture was cooled to room temperature before being quenched with H2O (10 mL) and EtOAc (20 mL). The two layers were separated, and the aqueous layer was extracted with EtOAc (2 x 20 mL). The combined organic extracts were washed with H2O (2 x 10 mL) and saturated aqueous NaCl (10 mL), dried over MgSO4, and concentrated in vacuo. The residue was then purified by flash column chromatography (0-3 % MeOH-CH2Cl2 gradient elution) to afford the desired (iSr,5)-(l-{4'-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2'-fluoro- biphenyl-4-yl}-2-cyano-ethyl)-carbamic acid tert-hvttyl ester (intermediate 18, 139 mg, 59% yield) as a yellow oil, which solidified upon standing at room temperature in vacuo.
A solution of intermediate 18 (85 mg, 0.17 mmol) in EtOAc (1.0 mL) was treated with a solution of 4 N HCl in 1,4-dioxane (3.0 mL), and the resulting reaction mixture was stirred at room temperature for 30 min. When TLC and HPLC showed that the reaction was complete, the solvents were removed in vacuo, to yield the monohydrochloride salt of the desired (S,S)-N-{3- [4'-(l-amino-2-cyano-ethyl)-2-fluoro-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide (HCl salt of compound 112, 64.0 mg, 94.1% yield) as yellow solids. LCMS (EI): mle 397 (M+ + H).
Example 7 - Synthesis of Compounds 113 - 116 Synthesis of compound 113
Scheme 6 illustrates the synthesis of compound 113 starting from 4-bromostyrene 19. Scheme 6
To a solution of 4-bromostyrene 19 (5.0Og, 26.77 mmol) in CH2Cl2 (130.0 mL) was added 4-niethylmorphorine JV-oxide (NMO, 12.9Og, 107.08 mmol, anhydrous) and (IS, 2S)-(+)- [l,2-(cyclohexanodiamino-N,N'-bis(3,5-di-t-butyl-salicylidene)] manganese(III) chloride
(850 mg, 1.34 mmol). The solution was cooled to -78 0C, then, rø-CPBA (7.40 g, 42.83 mmol) was added in four portions, one every 10 min. The mixture was stirred at -78 0C for 2 hr. The reaction was quenched by addition of aqueous sodium thiosulfate (Na2S2O3, 10.0 g in 30 mL H2O), then the cooling bath was removed and H2O (70 mL) and IN sodium hydroxide (NaOH, 60 mL) were added. The aqueous phase was extracted with CH2Cl2 (30 mL x 3), and the organic extracts were dried over Na2SO4. Flash column chromatography (4:100 diethyl ether (Et2O)/hexane) yielded 5.20 g epoxide 20.
To a solution of epoxide 20 (500 mg, 2.51 mmol) in EtOH (2.0 mL) at 55 0C was added NaCN (194 mg, 3.77 mmol 1 mL in H2O) and acetic acid (AcOH, 166 mg, 2.76 mmol in 1 mL H2O) over 10 min. The mixture was stirred at 55-60 0C for 2 hr. The solvents were removed in vacuo and the residue was purified by flash column chromatography (25:75 to 35:65 EtOAc/ Hexane) to yield 53 mg intermediate 21.
A suspension of intermediate 21 (1 eq.), boronate 10 (1 eq.), PdCl2(dppf)2 (0.05 eq.), and K2CO3 (4 eq.) in 3:1 :1 dioxane/EtOH/H2O was degassed with argon. The mixture was stirred at 90 °C for 4.5 hr. The solvent was removed in vacuo and the residue was separated by flash column chromatography (5:100 MeOH/CH2Cl2) to yield compound 113 (136 mg, 85.5 % yield). LCMS (ESI): m/e 398 (M+H)+.
Synthesis of compound 114
Compound 114 was prepared according to the synthesis described for compound 113, above, using (IR, 2R)-(+)-[l,2-(cyclohexanodiamino-N,N'-bis(3,5-di-t-butyl-salicylidene)] manganese(III) chloride, to yield 113 mg product (83% yield). LCMS (ESI): m/e 398 (M+H)+.
Synthesis of compound 115
Compound 115 was prepared according to the synthesis described for compound 113, above, using (IR, 2R)-(+)-[l,2-(cyclohexanodiamino-N,N'-bis(3,5-di-t-butyl-salicylidene)] manganese(III) chloride, to yield 34 mg product (69% yield). LCMS (ESI): m/e 371 (M+H-CN)+.
Synthesis of compound 116
Compound 116 was prepared according to the synthesis described for compound 113, above, starting from 4-bromo-2-vinylpyridine and using (IR, 2R)-(+)-[l,2- (cyclohexanodiamino-N,N'-bis(3,5-di-t-butyl-salicylidene)] manganese(IH) chloride, to yield 140 mg product (91% yield). LCMS (ESI): m/e 399 (M+H)+.
Example 8 - Synthesis of Compound 117
Scheme 7
To a solution of alcohol 22 (5.6 g, 27.85 mmol), imidazole (3.8 g, 55.7 mmol) and DMAP (cat.) in DMF (55 mL) was added (l,l-dimethylethyl)-diphenylsilyl chloride (TBDPSCl, 7.2 mL, 27.85 mmol) at 0 0C and the mixture was stirred at ambient temperature for 72 hr. The reaction was quenched with ice water (50 mL) and extracted with ether (4 x 50 mL). The combined etheral layer was washed with H2O (4 x 100 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. Flash column chromatography (2% EtOAc in hexanes) yielded 10.6 g of silylated alcohol 23.
To a solution of alcohol 23 (10.5 g, 24 mmol) in THF (50 mL) was added H-BuLi (2.5M in hexane, 11.5 mL, 28.8 mmol) at -78 0C and the mixture was stirred for 1 hr before the addition of trimethyl borate (3.54 mL, 31.2 mmol). The solution was stirred overnight at ambient temperature. The reaction mixture was quenched with IM KHSO4 (25 mL) and extracted with CH2Cl2 (3 x 50 mL). The organic layer was washed with brine (3 x 100 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. Flash column chromatography (25% EtOAc in hexanes) yielded 5 g of boronic acid 24 as mixture of acid and cyclic anhydrides.
To a mixture of boronic acid 24 (4.7 g, 11.68 mmol), oxazolidinone intermediate 1 (4 g, 10.6 mmol), K2CO3 (4.4 g, 31.8 mmol) and Pd(PPh3)4 (0.613 g, 5 mol%) was added toluene (90 mL), EtOH (30 mL) and H2O (30 mL). The reaction mixture was refluxed overnight under argon atmosphere, concentrated in vacuo and redissolved in CH2Cl2 (100 mL). The organic phase was washed with brine solution (2 x 100 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. To a solution of this crude material in THF (70 mL) was added
tetrabutylammonium fluoride (TBAF, 20 mL, 20 mmol) and the reaction mixture was stirred overnight at ambient temperature. The reaction mixture was concentrated in vacuo and washed with H2O (4 x 100 mL) to yield 3.5 g of biaryl intermediate 25. LCMS (ESI): m/e 373 (M+H).
To a solution of biaryl intermediate 25 (1 g, 2.7 mmol) in CH2Cl2 (15 mL), DMF (4 mL) and Hunig's base (0.75 mL, 4.05 mmol) was added MsCl (0.315 mL, 2.7 mmol) at 0 0C. After 2 hr the reaction mixture was poured into CH2Cl2 (150 mL) and the organic layer was washed with H2O (3 x 100 mL), dried, and concentrated in vacuo to yield crude mesylate. A mixture of crude mesylate (153 mg, 0.34 mmol) and NaCN (66 mg, 1.36 mmol) was heated in DMF (5 mL) at 80 0C for 2h. The reaction mixture was concentrated and purified by flash chromatography (20: 1 CH2Cl2MeOH) to yield 106 mg of compound 117. LCMS (ESI): m/e 382 (M+H).
Example 9 - Synthesis of Compounds 118 and 119
Scheme 8
119 A solution of 0.060 g (0.17 mmol) of amine 5 and 0.098 g (0.67 mmol) of dimethyl N- cyanodithioiminocarbonate 26 in 2 ml of DMF was stirred at 25 0C for 2 hr. The solvent was removed in vacuo and the residue was purified by preparative TLC to give 0.030 g of the desired compound 118. MS: 456 (M+H).
A solution of 0.060 g (0.22 mmol) of compound 118 and 1.5 ml of methylamine (MeNH2, 40% solution in H2O) in 6 ml of DMF was stirred at 25 0C for 20 hr. The solvents were removed in vacuo, and the residue was purified by preparative TLC to give 0.065 g of the desired compound 119. MS: 439 (M+H).
Example 10 - Synthesis of Compound 121
Scheme 9
A solution of the N-{3-[4-(6-aminomethyl-pyridin-3-yl)-3-fluoro-phenyl]-2-oxo- oxazolidin-5-ylmethyl}-acetamide (amine 27, 112 mg, 0.31 mmol) in DMF (4.0 mL) was treated with S-methyl-N-cyano-N'-methyl-carbamimidothioate 28 (61 mg, 0.47 mmol) at room temperature, and the resulting reaction mixture was warmed to 100 0C and stirred for 2 hr. When TLC and MS showed the reaction was complete, the reaction mixture was concentrated in vacuo, and the residue was directly purified by column chromatography (0-5% MeOH/CH2Cl2 gradient elution) to afford the desired f5)ξ)-N-{3-[4-(6-N-(N-methyl-N'-cyano)guanylaminomethyl- pyridin-3-yl)-3-fluoro-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide (compoundl21, 5.2 mg, 3.8 % yield) as off-white solids. LCMS (EI): m/e 440 (M + H).
Example 11 - Synthesis of Compound 122
Scheme 10
122
To a suspension of alcohol 29 (5 g, 14.84 mmol) in CH2Cl2 (80 niL) was added Et3N (2.5 mL, 17.8 mmol) and MsCl (1.4 mL, 17.8 mmol) at 0 0C and the solution was stirred for 1 hr. The reaction mixture was poured into brine (100 mL) and the aqueous was extracted with CH2Cl2 (2 x 50 mL). The combined organic layer was washed with brine (3 x 100 mL), dried over anhydrous Na2SO4, and concentrated to yield mesylate 30. To this was added NaN3 (2 g, 29.7 mmol) and DMF (50 mL) and the mixture was heated to 80 0C overnight. The solution was poured into a mixture of EtOAc (150 mL) and H2O (100 mL). The organic layer was separated and the aqueous was extracted with EtOAc (3 x 50 mL). The combined organic layers were washed with brine (1 x 150 mL), dried over anhydrous Na2SO4, and concentrated to yield 5.4 g of the azide 31.
A solution of azide 31 (5.4 g, 14.84 mmol) and trimethylsilyl acetylene (TMS acetylene, 10.48 mL, 74.2 mmol) in DMF (20 mL) was heated to 90 0C for 12 hr. The reaction mixture was concentrated in vacuo, treated with TBAF (60 mL, IM in THF) and AcOH (2 mL, 29.7 mmol), and stirred at ambient temperature for 12 hr. The solution was concentrated and poured into a mixture of saturated NH4Cl (50 mL), EtOAc (150 mL) and brine (50 mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (3 x 50 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated, and the solid thus obtained was washed with H2O (5 x 200 mL) to yield 5.7 g of tetrazole derivative 32. LCMS (ESI): m/e 389 (M+H+). To a mixture of tetrazole 32 (5.7 g, 14.84 mmol), boronic acid 33 (2.9 g, 19.29 mmol),
K2CO3 (6.0 g, 44.52 mmol) and Pd(PPh3)4 (857 mg, 5 mol %) was added toluene (120 mL), EtOH (40 mL) and H2O (40 mL). The reaction mixture was degassed and flushed with argon and refluxed for 4 hr. The solvent was concentrated in vacuo and the residue thus obtained was added to H2O (2000 mL). The pale yellow solid was filtered, and dried at 40 0C in vacuo to yield 4.76 g of alcohol 34. LCMS (ESI): m/e 369 (M+H+).
To a solution of alcohol 34 (4.6 g, 12.5 mmol) and Hunig's base (6.4 mL, 38.75 mmol) in DMF (40 mL) and CH2Cl2 (30 mL) was added MsCl (2.9 mL, 37.5 mmol) at 0 0C and the resulting solution was stirred at ambient temperature for 3 h. The solution was concentrated to remove the CH2Cl2 and poured into H2O (1000 mL). The pale yellow solid was filtered and successively washed with H2O (5 x 200 mL), 10% EtOAc in hexanes (5 x 100 mL) and 50% ether in hexanes (5 x 100 mL). The resulting solid was dried at 40 0C in vacuo to yield 4.5 g of chloride 35. LCMS (ESI): m/e 387 (M+H+).
A mixture of chloride 35 (100 mg, 0.258 mmol), 3-aminopropionitrile (0.330 mL, 4.48 mmol) and Hunig's base (0.400 mL) in DMF (2 mL) was stirred at ambient temperature overnight. To the reaction mixture was added CH2Cl2 (30 mL), MeOH (3 mL) and brine (30 mL). The aqueous was extracted with 10% MeOH-CH2Cl2 (2 x 30 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (30:1:0.01 CH2Cl2/MeOH/NH4OH) to yield 55 mg of compound 122. LCMS (ESI): m/e 443 (M+H+Na+).
Example 12 - Synthesis of Compound 123 Scheme 11
NC
Azide 37 and chloride 38 were synthesized from alcohol 36 using Mitsunobu conditions. Formation of the acid HN3 (produced by NaN3 and HCl) produces chloride ions which can also act as nucleophiles, leading to the inseparable mixture of 37 and 38. When the mixture was coupled with boronate 10 under conditions described in Example 7, both starting materials underwent an elimination reaction to form a double bond and yielded the coupling product as an inseparable 1 :3 Z/E mixture.
Example 13 - Synthesis of Compound 129
Scheme 12
129
Synthesis of chloride 39 Alcohol 2 (3.0 g, 8.4 mmol) 51 was dissolved in CH2Cl2 (20 niL) and Hunig's base (2 niL). MsCl (1.4 mL, 12.6 mmol) was added dropwise and the resulting solution stirred at room temperature for 4 hr. The mixture was poured into 100 mL saturated aqueous NaHCO3 and extracted with CH2Cl2 (3 x 50 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated to give 3.9 g of an oily yellow solid. The crude material was purified by silica gel chromatography to give chloride 39 as an off-white solid (2.7 g, 7.2 mmol). LCMS (ESI): m/z 377 (M + H)+, 418 (M + CH3CN + H)+, 440 (M + CH3CN + Na)+.
Synthesis of compound 129
To a solution of chloride 39 (753 mg, 2 mmol) and Hunig's base (0.4 mL, 2.3 mmol) in DMF (5 mL) was added a solution of propargyl amine (220 mg, 4 mmol) in DMF (1 mL). After stirring at room temperature for 24 h, the DMF was removed in vacuo. The resulting residue was dissolved in THF (15 mL) and H2O (3 mL). K2CO3 (345 mg, 2.5 mmol) and di-tert-butyl dicarbonate (436 mg, 2 mmol) were added, and the reaction mixture was stirred at room temperature for 2 hr. The THF was removed in vacuo, 50 mL of EtOAc was added, and the solution was washed with H2O, dried over MgSO4 and concentrated in vacuo. The crude product was purified by preparative TLC (15:1:0.05 CH2Cl2MeOH/ NH3H2O) to afford 930 mg of Boc- protected amine 40 (94% yield). MS (ESI): 439.9, 518.0 (M+Na)+, 481.1, 559.1 (100%).
To a solution of Boc-protected amine 40 (140 mg, 0.28 mmol) in CH2Cl2 (5 niL) and MeOH (1 niL) was added 2 ml of 4.0 M HCl in dioxane. After stirring at room temperature for 4 hr, the reaction was concentrated in vacuo and washed with EtOAc/MeOH to give 115 mg of the monohydrochloride salt of compound 129 (94% yield). MS (ESI): 359.0 (100%), 396.1(M+H)+.
Example 14 - Synthesis of Compound 131
A mixture of amine 5 (179 mg, 0.5 mmol), 3-bromo-propene (60 mg, 0.5 mmol), Hunig's base (0.17 mL, 1 mmol) and KI (10 mg) in DMF (5 mL) was heated at 70 0C for 12 hr. The reaction was diluted with EtOAc (25 mL), washed with H2O, dried over MgSO4 and concentrated in vacuo. The crude product was purified by preparative TLC (15:1 :0.05 CH2Cl2/MeOH/MΪ3.H2O) to afford 48 mg of desired compound 131 (42% yield). MS (ESI): 398.1 (100%, (M+H)+).
Example 15 - Synthesis of Compounds 133 and 134
A mixture of amine 5 (179 mg, 0.5 mmol), 4-bromo-l-butene (68 mg, 0.5 mmol), Hunig's base (0.17 mL, 1 mmol) and KI (10 mg) in DMF (5 mL) was heated at 70 0C for 12 hr. The reaction was neutralized with 2N NaOH and then EtOAc (25 mL) was added. The organic phase was separated, washed with H2O, dried over MgSO4 and concentrated in vacuo. The crude product was purified by preparative TLC (15:1:0.05 CH2Cl2/ MeOH/NH3.H2O) to afford 106 mg of compound 133 (52% yield) and 106 mg of compound 134 (15% yield). MS (ESI) of 133: 412.0 (100%, (M+H)+). MS (ESI) of 134: 466.2 (100%, (M+H)+).
Example 16 - Synthesis of Compound 135 A mixture of amine 5 (629 mg, 1.7 mmol), but-4-ynyl tosylate (395 mg, 1.7 mmol) and
Hunig's base (0.6 mL, 3.5 mmol) in DMF was heated at 50 0C for 16 hr. The reaction concentrated in vacuo and the residue was purified by preparative TLC (15:1:0.05 CH2Cl2/MeOH/NH3.H2O) to afford 220 mg of desired compound 135 (31% yield). MS (ESI): 410.1(100%, (M+H)+), 432.1 (M+Na)+.
Example 17 - Synthesis of Compound 136
A mixture of amine 5 (378 mg, 1.1 mmol), pent-5-ynyl tosylate (252 mg, 1.1 mmol) and Hunig's base (0.35 mL, 2 mmol) in DMF (6 mL) was heated at 50 0C for 8 hr. The reaction concentrated in vacuo and the residue was purified by preparative TLC (10:1 :0.05 CH2Cl2/MeOH/NΗ3.H2O) to afford 140 mg of desired compound 136 (41% yield). MS (ESI): 424.1(100%, (M+H)+), 446.1 (M+Na)+.
Example 18 - Synthesis of Compound 137
A mixture of amine 5 (179 mg, 0.5 mmol), 5-bromo-l-pentene (75 mg, 0.5 mmol), KI (10 mg) and Hunig's base (0.17 mL, 1 mmol) in DMF (2 mL) was heated at 70 0C for 12 hr. The reaction concentrated in vacuo and the residue was purified by preparative TLC (15:1:0.05 CH2Cl2/MeOH/NH3.H2O) to afford 80 mg of desired compound 137 (38% yield). MS (ESI): 426.1(100%, (MH-H)+).
Example 19 - Synthesis of Compound 138
A mixture of amine 5 (179 mg, 0.5 mmol), crotyl bromide (68 mg, 0.5 mmol), KI (10 mg) and Hunig's base (0.17 mL, 1 mmol) in DMF (2 mL) was heated at 70 0C for 12 hr. The reaction concentrated in vacuo and the residue was purified by preparative TLC (15:1:0.05 CH2Cl2/MeOH/NH3.H2O) to afford 38 mg of desired compound 138 (18% yield). MS (ESI): 412.1(100%, (M+H)+).
Example 20 - Synthesis of Compound 139
A mixture of amine 5 (179 mg, 0.5 mmol), l-bromo-but-2-yne (67 mg, 0.5 mmol), KI (10 mg) and Hunig's base (0.17 mL, 1 mmol) in DMF (2 mL) was heated at 70 0C for 5 hr. The reaction concentrated in vacuo and the residue was purified by preparative TLC (15:1:0.05 CH2Cl2/MeOH/NH3.H2O) to afford 45 mg of desired compound 139 (22% yield). MS (ESI): 410.1(100%, (MH-H)+), 432.0 (MH-Na)+.
Example 21 - Synthesis of Compound 144 A mixture of amine 5 (357 mg, 1 mmol), l-bromo-but-3-en-2-ol (151 mg, 1 mmol), KI
(20 mg) and Hunig's base (0.26 mL, 1.5 mmol) in DMF (3 mL) was heated at 700C for 24 h.
The reaction concentrated in vacuo and the residue was purified by preparative TLC (10:1 :0.05 CH2Cl2/MeOH/NH3.H2O) to afford 65 mg of crude compound 144. The crude product was dissolved in THF (5 mL) and H2O (1 mL), and K2CO3 (41 mg, 0.3 mrnol) and di-tert-butyl dicarbonate (44 mg, 0.2 mmol) were then added. The reaction was stirred at room temperature for 2 hr. The THF was removed in vacuo and 50 mL of EtOAc was added. The solution was washed with H2O, dried over MgSO4 and concentrated. The crude product was purified by preparative TLC (15:1:0.05 CH2Cl2/ MeOHMB3 H2O) to afford 54 mg of Boc-protected amine. This Boc-protected amine was dissolved in CH2Cl2 (5 mL) and MeOH (1 mL), and 1 mL of 4.0 M HCl in dioxane was added. After stirring at room temperature for 4 hr, the reaction was concentrated and washed with EtOAc/MeOH to give 45 mg of the monohydrochloride salt of compound 144 (10% yield). MS (ESI): 428.1 (100%, (M+H)+).
Example 22 - Synthesis of Compound 145
To a solution of chloride 35 (synthesized according to the procedure in Example 11) (580 mg, 1.5 mmol) and Hunig's base (0.7 mL, 4.0 mmol) in DMF (5 mL) was added a solution of propargyl amine (275 mg, 5 mmol) in DMF (1 mL). After stirring at room temperature for 20 hr, the DMF was removed in vacuo. The resulting residue was dissolved in THF (10 mL) and H2O (2 mL), and K2CO3 (276 mg, 2 mmol) and di-tert-butyl dicarbonate (324 mg, 1.5 mmol) were then added. The reaction was stirred at room temperature for 2 hr, then the THF was removed in vacuo. 50 mL of EtOAc was added and the solution was washed with H2O, dried over MgSO4 and concentrated. The crude product was purified by preparative TLC (15:1 :0.05 CH2Cl2ZMeOHMH3 H2O) to afford 670 mg of Boc-protected amine (88% yield). MS (ESI): 450.0, 491.0(100%), 506.2 (M+H)+.
To a solution of the Boc-protected amine (35 mg, 0.07 mmol) in CH2Cl2 (4 mL) and MeOH (0.5 mL) was added 0.5 ml of 4.0 M HCl in dioxane. After stirring at room temperature for 12 hr, the reaction was concentrated and washed with EtOAc/MeOH to give 26 mg of the monohydrochloride salt of compound 145 (93% yield). MS (ESI): 369.0 (100%), 410.0 (M+H)+).
Example 23 - Synthesis of Compound 148
Scheme 13
A suspension of the monohydrochloride salt of 5-aminomethyl-3-(4'-chloromethyl-2- fluoro-biphenyl-4-yl)-oxazolidin-2-one (HCl salt of amine 41, 180 mg, 0.54 mmol) in CH2Cl2 (10.0 mL) was treated with difluoro-acetic acid 42 (118 mg, 1.2 mmol), monohydrochloride salt of EDCI (230 mg, 1.2 mmol) and Hunig's base (209 mg, 1.62 mmol) at room temperature, and the resulting reaction mixture was stirred at room temperature for 24 hr. When TLC and MS showed the reaction was complete, the reaction mixture was concentrated in vacuo, and the residue was directly purified by flash column chromatography (0-5% MeOH/CH2Cl2 gradient elution) to afford the desired N-[3-(4'-Chloromethyl-2-fluoro-biphenyl-4-yl)-2-oxo-oxazolidin-5- ylmethyl]-2,2-difluoro-acetamide (acetamide 43, 100 mg, 44.5% yield ) as yellow solids. LCMS (EI): mle 413 (M+ + H).
A solution of acetamide 43 (190 mg, 0.46 mmol) in DMF (10.0 mL) was treated with propargylamine (55 mg, 1.0 mmol) and Hunig's base (71.3 mg, 0.55 mmol) at room temperature, and the resulting reaction mixture was stirred at room temperature for 24 hr. When TLC and MS showed the reaction was complete, H2O and (BOC)2O were added and the reaction mixture was stirred at room temperature for 2 hr. The reaction solution was concentrated in vacuo, and the residue was directly purified by flash column chromatography (0-5% MeOH/CH2Cl2 gradient
elution) to afford the desired (4'-{5-[(2,2-Difluoro-acetylamino)-metliyl]-2-oxo-oxazolidin-3- yl}-2'-fluoro-biphenyl-4-ylmethyl)-piOp-2-ynyl-carbamic acid tert-butyl ester (Boc-protected amine 44). (194 mg, 97.8% yield ) as off-white solids. LCMS (EI): mle 432 (M+ + H).
A solution of Boc-protected amine 44 in CH2Cl2 was treated with excess 4.0 M HCl in dioxane to afford the HCl salt of the desired 2,2-difluoro-N-[3-(2-fluoro-4'-prop-2- ynylaminomethyl-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (compound 148).
Example 24 - Synthesis of Compounds 149 and 150
Scheme 14
39 148
m-CPBA
149
A mixture of chloride 39 (149 mg, 0.395 mmol), 2-(methylthio)ethylamine (148 mg, 1.626 mmol), K2CO3 (55 mg, 0.395 mmol) and KI (5 mg) in DMF (2 mL) was heated at 70 0C for 2 hr. The reaction was diluted with EtOAc (25 mL), washed with H2O, dried over MgSO4 and concentrated in vacuo. The crude product was purified by preparative TLC (15:1:0.05 CH2Cl2/MeOH/NH3.H2O) to afford 138 mg of desired compound 149 (81% yield). MS (ESI): 432.1(100%, (M+H)+), 573.1 (M+Na)+. 1HNMR (300 MHz, CDCl3 ): δ 7.54-7.38 (m, 6H), 7.27 (m, IH), 6.05 (t, J= 6Hz, IH), 4.81 (m, IH), 4.09 (t, J= 9Hz, IH), 3.86 (s, 2H), 3.81 (dd, J= 7, 9Hz, IH), 3.78-3.61(m, 2H), 2.86 (t, J= 7Hz, 2H), 2.70 (t, J= 7Hz, 2H), 2.09 (s, 3H), 2.04 (s, 3H) . A mixture of compound 149 (58 mg 0.13 mmol) and λn-CPBA (70%, 33 mg, 0.13 mmol) in CH2Cl2 was stirred at room temperature for 1 hr. The CH2Cl2 solution was washed with brine, dried over MgSO4 and concentrated in vacuo. The crude product was purified by preparative TLC (15:1:0.05 CH2Cl2/MeOH/NH3.H2O) to afford 42 mg of compound 150 (72% yield). MS (ESI): 448.1 (100%, (M+H)+), 470.1 (M+Na)+.
Example 25 - Synthesis of Compounds 151 and 152
A mixture of amine 5 (357 mg, 1 mmol), 3-methylthiopropyl methanesulfonate (184 mg, 1 mmol), Hunig's base (0.21 mL, 1.2 mmol) and KI (10 mg) in DMF (5 mL) was heated at 70 0C for 48 hr. The reaction was neutralized with 2N NaOH and then EtOAc (25 mL) was added. The organic phase was separated, washed with H2O, dried over MgSO4 and concentrated in vacuo. The crude product was purified by preparative TLC (15:1:0.05 CH2Cl2/MeOH/NH3.H2O) to afford 70 mg of compound 151 (16% yield) and 25 mg of compound 152 (9% yield). MS (ESI) of 151: 446.1 (100%, (M+H)+). MS (ESI) of 152: 534.1 (100%, (M+H)+).
Example 26 - Synthesis of Compound 153
Scheme 15
A mixture of methylamine 45 (111 mg, 0.3 mmol), methylthioethyl chloride (55 mg, 0.5 mmol), K2CO3 (41 mg, 0.3 mmol) and KI (10 mg) in DMF (2 mL) was heated at 70 0C for 12 hr. The reaction was diluted with EtOAc and the organic phase was separated, washed with H2O, dried over MgSO4 and concentrated in vacuo. The crude product was purified by preparative TLC (25:1:0.05 CH2Cl2/MeOH/NH3.H2O) to afford 61 mg of compound 153 (46% yield). MS (ESI): 446.1 (100%, (M+H)+).
Example 27 - Synthesis of Compound 154
SchemeJ6
A mixture of chloride 46 (109 mg, 0.25 mmol), 2-(methylthio)ethylamine (94 mg, 1.25 mmol), K2CO3 (41 mg, 0.30 mmol) and KI (5 mg) in DMF (2 mL) was heated at 70 0C for 1 hr. The reaction was diluted with EtOAc (25 mL), washed with H2O, dried over MgSO4 and concentrated in vacuo. The crude product was purified by preparative TLC (15:1:0.05
CH2Cl2/MeOH/NH3.H2O) to afford 55 mg of desired compound 154 (51% yield). MS (ESI): 433.1(100%, (M+H)+).
Example 28 - Alternative Synthesis of Compound 133 Scheme 17
133, Mono Hydrochloride
To a solution of chloride 39 (8.28 g, 22.0 mmol) and l-amino-3-butene hydrochloride (4.73 g, 44.0 mmol) in DMF (50 mL) was added Hunig's base (12.4 mL, 71.5 mmol). After stirring at room temperature for 3 days, DMF was removed in vaccum. The resulted residue was dissolved in a mixed solvent of THF (150 mL) and H2O (30 mL), and K2CO3 (6.9 g, 50.0 mmol) and di-tert-butyl dicarbonate (5.0 g, 23.1 mmol) were added. The reaction was stirred at room temperature for 2 hr, then THF was removed in vacuum. EtOAc (150 mL) was added and the solution was washed with 0.5 N aqueous HCl, 10% aqueous NaHCO3, dried over MgSO4 and concentrated. The crude product was purified by column chromatography (25 : 1 :0.05 / CH2Cl2:MeOH:NH3 H2O) to afford Boc-protected amine 47 (6.75 g, 60 % yield).
To a solution of Boc-protected amine 47 (6.75 g, 13.2 mmol) in CH2Cl2 (100 mL) and MeOH (10 mL) was added 20 ml of HCl solution (4.0 M in dioxane). After stirring at room temperature for 12 hr, the reaction was concentrated, washed with CH3CNZMeOH (95:5) to give the monohydrochloride salt of compound 133 (4.68 g, 79 % yield). MS (ESI): 412.2 (100%, (M+H)+).
INCORPORATION BY REFERENCE
The entire disclosure of each of the patent documents and scientific articles referred to herein is incorporated by reference for all purposes.
EQUIVALENTS
The invention can be embodied in other specific forms without departing from the spirit or essential characteristics thereof. The foregoing embodiments are therefore to be considered in all respects illustrative rather than limiting on the invention described herein. Scope of the invention is thus indicated by the appended claims rather than by the foregoing description, and all changes that come within the meaning and range of equivalency of the claims are intended to be embraced therein.