SUBSTITUTED 2 , 3 , 5-TR FLUORPHENYL OXAZOLIDINONES FOR USE AS ANTIBACTERIAL AGENTS FILED OF THE INVENTION The present invention relates to substituted trifluorphenyl oxazolidinones and 5 to the processe for the synthesis of the same. The compounds of the present invention are useful antimicrobial agents, effective against a number of human and veterinary pathogens, including gram-positive aerobic bacteria such as multiply-resistant staphylococci, streptococci, and enterococci as well as anaerobic organisms such as bacteroides spp. and clostridia spp. species, and acid-fast organisms such as 10 Mycobacterium tuberculosis, Mycobacterium avium and Mycobacterium spp. This invention also relates to a novel combination therapy for treating infective diseases caused by bacteria in human or animal.
BACKGROUND OF THE INVENTION 15 Due to ever-increasing antibiotic resistance, structurally novel antibacterials with a new mode of action have become increasingly important in the treatment of bacterial infections. Among newer antibacterial agents, oxazolidinone compounds are the most recent synthetic class of antimicrobials active against a number of pathogenic microorganisms, including pathogens resistant to other clinically useful antibiotics. 20 It is also known that as a chemical compound class, oxazolidinones generally inhibit to some extent monoamine oxidase (MAO), the enzyme responsible for preventing acute blood pressure elevation by the endogenous and dietary amine, tyramine, and other sympathomimetic amines. Accordingly, there is a demand to discover oxazolidinone antibiotics that possess minimum MAO inhibitory activity. 25 The present invention provides sulfur-containing heterocyclic trifluorphenyl oxazolidinones of formula I. These compounds has unexpectedly weak MAO inhibitory activity, which indicates that these compounds possess the capacity to minimize or eliminate potential drug-drug interactions since strong inhibition of monoamine oxidase can result in altered clearance rates for other compounds 30 normally metabolized by it. Many classes of antibiotic compounds, including quinolones, have been described for the treatment of infectious diseases, particularly bacterial infections. The development of bacterial resistance to currently available antibacterial agents is a
growing global health problem. Of particular concern are infections caused by multidrug-resistant pathogens, for example, methicillin-resistant Staphylococcus aureus (MRS A), vancomycin-resistant Enterococci (VRE), glycopeptide-intermediate Staphylococcus aureus (GISA), and vancomycin-intermediate Staphylococcus aureus (VISA). The reduced susceptibility to known antibacterial agents creating an ongoing need for developing effective therapeutic measures. The present invention may be used in a combination therapy for treating bacterial infections which comprises administration to a mammal a compound of present invention and a second antibiotic agent to achieve broad coverage and synergistic interactions. International Patent Publication No. WO 03/063862 discloses that a wide variety of oxazolidinones may be used in combination with B vitamins in the treatment of bacterial infections. US Patent 6,239,152 discloses oxazolidinones and methods for their synthesis. US Patent No 6,605,609 discloses a thizaine oxazolidinone useful as antimicrobial agent and a new antimicrobial combination therapy for infective diseases. US Patent No 5,700,799 discloses oxazolidinone derivatives possessing a substituted diazine moiety bonded to an N-aryl ring. US Patent No 6,605,609 discloses thiopyran oxazolidinones useful as antimicrobial agents. US Patent No. 5,880,118 discloses substituted oxazine and thiazine oxazolidinone antimicrobials. US Patent No. 6,968,962 discloses phenyloxazolidinones having a C-C bond to 4-8 membered heterocyclic rings. US Patent No. 5,981,528 discloses antibiotic oxazolidinone derivatives. International Patent Publication No. WO 01/34128 A2 discloses admixture of linezolid and other antibacterial agents. International Patent Publication No. WO 03/093247 A discloses oxazolidinone derivatives for treating or preventing infectious disorder caused by bacteria in human or animal. International Patent Publication No. WO 03/0007870 A2 discloses certain substituted phenyl oxazolidinones and processes for the synthesis of the same. US Patents 4,382,892, 4,670,444, 4,670,444, 4,703,047, 4,758,567, 4,820,716,
4,889,857, 5,039,683, 5,077429, 5,262,417, and 5,385,906 disclose quinolone antibiotics that may be used in combination with the compounds of the present invention. None of the references cited above specifically contemplates the compound of the present invention, its combination therapy and its novel compositions.
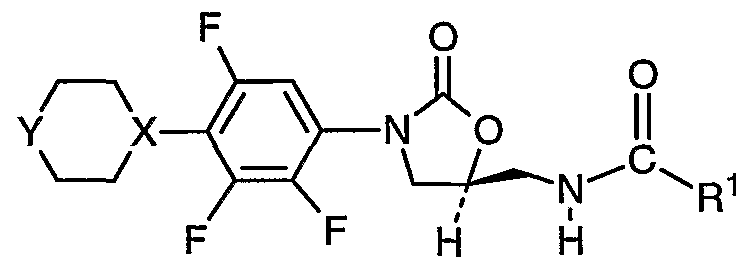
SUMMARY OF THE INVENTION The present invention provides sulfur-containing heterocyclic trifluorphenyl oxazolidinone compounds of formula I
I or its pharmaceutically acceptable salt or a prodrug thereof wherein X is -CH- or -N-, Y is -O-, -S(O)„-; or X is -N-, Y is HOCH
2C(=O)N-; Ri is -C
1-6 alkyl, -OC
1-6 alkyl, or -NHC
1-6 alkyl; and n is 0, 1 or 2. Specifically, R
1 is -CH
3, X is -N-, and Y is HOCH
2C(=O)N-. Specifically, R
1 is -CH
3, X is -N-, and Y is -O-. Specifically, R
1 is -CH
3, X is -N-, and Y is -SO
2-. These compounds have potent activity against a number of human and veterinary bacterial pathogens, and have excellent safety profiles. The present invention further provides a method for treating bacterial infections which comprises administration to a mammal being treated a pharmaceutically effective amount of the compound of formula I, either individually, or in combination with at least one second antibiotics. The present invention further provides compositions for treating bacterial infections wherein the compositions comprise a pharmaceutically effective amount of the compound of formula I and a pharmaceutically acceptable carrier. These and other aspects, advantages, and features of the invention will become apparent from the following detailed description of the invention
DETAILED DESCRIPTION OF THE INVENTION
Definitions
A "pharmaceutically acceptable carrier" refers to a carrier that is useful in preparing a pharmaceutical composition that is generally safe, non-toxic and neither biologically nor otherwise undesirable, and includes a carrier that is acceptable for veterinary use as well as human pharmaceutical use. "A pharmaceutically acceptable carrier" as used in the specification and claims includes both one and more than one such carrier. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable, non-toxic bases and acids. Pharmaceutically acceptable, non-toxic bases and acids include inorganic bases, inorganic acids, organic acids, and inorganic bases. Salts derived from inorganic bases include aluminum, ammonium, calcium, ferric, ferrous, lithium, magnesium, potassium, sodium, zinc, and the like. Salts derived from pharmaceutically acceptable, organic, non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, such as arginine, betaine, caffeine, choline, N, N-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2- dimethylamino-ethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N- ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, and the like. Salts derived from inorganic acids include salts of hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, mineral acids, sulfonic acids, phosphoric acid, phosphorous acid, and the like. Salts derived from pharmaceutically acceptable, organic, non-toxic acids include salts of C1-6 alkyl carboxylic acids, di- carboxylic acids, and tri-carboxylic acids such as acetic acid, propionic acid, fumaric acid, succinic acid, tartaric acid, maleic acid, adipic acid, and citric acid. Other salts may be derived from aryl and alkyl sulfonic acids such as toluene sulfonic acids and the like. Pharmaceutically acceptable salts may be obtained using standard procedures well known in the art, for example by reacting a sufficiently basic compound such as an amine with a suitable acid affording a physiologically acceptable anion. Alkali
metal (for example, sodium, potassium or lithium) or alkaline earth metal (for example calcium) salts of carboxylic acids can also be made. In addition to salt forms, the present invention provides compounds, which are in a prodrug form. The expression "prodrug" denotes a derivative of a known direct acting drug, which derivative has enhanced delivery characteristics and therapeutic value as compared to the drug, and is transformed into the active drug by an enzymatic, for example by hydrolysis in blood, or chemical process [see T. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A.C.S. Symposium Series; Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, (1987); Notari, R. E., "Theory and Practice of Prodrug Kinetics," Methods in Enzymology, 112:309-323 (1985); Bodor, N., "Novel Approaches in Prodrug Design," Drugs of the Future, 6(3): 165-182 (1981); and Bundgaard, H., "Design of Prodrugs: Bioreversible- Derivatives for Various Functional Groups and Chemical Entities," in Design of Prodrugs (H. Bundgaard, ed.), Elsevier, N.Y. (1985)]. The prodrug is formulated with the objective(s) of improved chemical stability, improved patient acceptance and compliance, improved bioavailability,prolonged duration of action, improved organ selectivity, improved formulation (e.g., increased hydrosolubility), and/or decreased side effects (e.g., toxicity). As used herein, a "prodrug" is any covalently bonded carrier that releases in vivo the active parent drug according to the Formula I when such prodrug is administered to the subject. Prodrugs of the compounds of Formula I are prepared by modifying functional groups present on the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound. Prodrugs include, but are not limited to, compounds derived from compounds of Formula I wherein hydroxy, amine or sulfhydryl groups are bonded to any group that, when administered to the subject, cleaves to form the free hydroxyl, amino or sulfhydryl group, respectively. Selected examples include, but are not limited to, biohydrolyzable amides and biohydrolyzable esters and biohydrolyzable carbamates, carbonates, acetate, formate and benzoate derivatives of alcohol and amine functional groups. Furthermore, prodrugs include compounds wherein an amino acid residue, or a polypeptide chain of two or more (e.g., two, three or four) amino acid residues is covalently joined through an amide or ester bond to a free amino, hydroxy or carboxylic acid group of compounds of Formula I and Formula π.
The amino acid residues include but are not limited to the 20 naturally occurring amino acids commonly designated by three letter symbols and also includes 4- hydroxyproline, hydroxylysine, demosine, isodemosine, 3-methylhistidine, norvalin, beta-alanine, gamma-aminobutyric acid, citrulline homocysteine, homoserine, ornithine and methionine sulfone. The term "treating" or "treatment" of a disease includes: (1) preventing the disease, i.e. causing the clinical symptoms of the disease not to develop in a mammal that may be exposed to or predisposed to the disease but does not yet experience or display symptoms of the disease, (2) inhibiting the disease, i.e., arresting or reducing the development of the disease or its clinical symptoms, or (3) relieving the disease, i.e., causing regression of the disease or its clinical symptoms. The term "therapeutically effective amount" refers to the amount of a compound that, when administered to a mammal for treating a disease, is sufficient to effect such treatment for the disease. The "therapeutically effective amount" will vary depending on the compound, the disease and its severity and the age, weight, etc., of the mammal to be treated. The term "combination therapy" refers to a treatment regimen wherein the compounds of the present invention and the second antibiotics are administered individually or together in such a way as to provide a beneficial effect from co-action of these therapeutic agents. Such beneficial effect can include, but is not limited to, pharmacokinetic or pharmacodynamic co-action of the therapeutic agents. Combination therapy can, for example, enable administration of a lower dose of one or both agents than would normally be administered during monotherapy, thus decreasing risk or incidence of adverse effects associated with higher doses. Alternatively, combination therapy can result in increased therapeutic effect at the normal dose of each agent in monotherapy. Alternatively, combination therapy can maximize the therapeutic effect at higher doses. The term "mammal" refers to human or warm-blooded animals including livestock and companion animals.
Methods for Preparation The compound of the present invention can be prepared according to the procedures described herein below. As shown in Scheme I, alkylation of 2,3,4,5- tetrafluoronitrobenzene a with thiomorpholine in an appropriate solvent such as
DMSO provides 4-(2,3,6-trifluoro-4-nitrophenyl)thiomorpholine b. The compound b is then treated with stannous chloride dihydrate (SnCl2) at a temperature in the range of about 25 C to about 100 C followed by protection of the aniline as the CBz- carbamate afforded the desired intermediate c.
SCHEME I
e Treatment of the Cbz carbamate c under conditions described in International Patent Publication No. WO 02/085849 A3 directly affords the desired trifluorinated oxazolidinones d. Compound d may be oxidized by the methods known to those skilled in the art to provide the corresponding sulf oxide and sulphone compounds. Alternatively, Cbz derivative c can first be converted to the corresponding carbamate derivatives e under conditions described in International Patent Publication No. WO 0232857 Al. When R
2 is tert-butyl, treatment with trifluoroacetic acid (neat or in dichloromethane) or hydrochloric acid in dioxane affords the corresponding 5- (aminomethyl)oxazolidinone intermediate which can be reacted with a variety of acetylating agents to provide the targeted compounds d. As shown in Scheme π, the starting material 2,3,5-trifluoroaniline a is converted to the protected silylated derivative b as described, for example, in Grega, K. C; et al. in J. Org. Chem. 1995, 60, 5255-5261 and Hutchinson, D. K.; et al. PCT Int. Appl. WO 9709328. It will be obvious to one skilled in the art that variations in the silyl protecting group are possible. Alternatively, the aniline a can be converted to
the protected dimethylpyrrole derivative c. Treatment of either b or c with n-BuLi in THF affords an intermediate lithiated phenyl ring which can be added to tetrahydrothiopyran-4-one (d) to give an intermediate adduct. The silyl protecting group can be removed by stirring with methanolic potassium carbonate or the like. Subsequent reaction with an appropriate alkyl chloroformate then affords the carbamate e. In the case of the dimethylpyrrole-protected intermediate adduct, deprotection can be achieved by treatment with hydroxylamine in aqueous ethanol. Again, subsequent reaction with an appropriate alkyl chloroformate then affords the carbamate e. Closely related chemistry pertinent to this coupling chemistry can be found in Herrinton, P. M.; et al. Org. Process Res. Develop. 2001, 5, 80-83 and in Poel, T. J.; et al. PCT Int. Appl. WO 9929688. Dehydration of the alcohol moiety of e, as described in Gage, J. R.; et al. Tetrahedron Lett. 2000, 41, 4301-4305, or variations thereof, then provides the unsaturated dihydrothiopyran intermediate f. Further transformation of intermediate f to advanced tetrahydrothiopyran intermediate g can be conducted as described in the above references. Conversion of g to the desired tetrahydrothiopyran examples h is accomplished by methods described for compounds in Schemes I and II and also as outlined in the above references. The unsaturated dihydrothiopyran intermediate f can also be elaborated to the dihydrothiopyran oxazolidinone derivative i. The double bond of i can be reduced as described in the above references to provide the corresponding tetrahydrothiopyran oxazolidinones h. The sulfur atom oxidation state of f, g, h and i can be controlled as described in the above references to give sulfide (n = 0), sulf oxide (n = 1) or sulfone (n = 2) derivatives. SCHEME H
Scheme HI illustrates the preparation of compounds wherein Y is oxygen and X is nitrogen. Alkylation of 2,3,4,5-tetrafluoronitrobenzene a with morpholine in an appropriate solvent such as DMSO provides 4-(2,3,6-trifluoro-4- nitrophenyl)morpholine b. The compound b is then treated with stannous chloride dihydrate (SnCl2) at a temperature in the range of about 25 C to about 100 C followed by protection of the aniline as the CBz-carbamate afforded the desired intermediate c. Alternatively, the nitro group of b can be reduced by hydrogenation with hydrogen gas in the presence of a suitable palladium catalyst such as 5-10% palladium on carbon. It will be apparent to one skilled in the art that these reduction conditions are merely representative and that other variations are possible.
SCHEME HI
k I When Y is oxygen, and X is carbon, the compounds may be prepared according to scheme IV. As shown in Scheme IV, the starting material 2,3,5- trifluoroaniline a is converted to the protected silylated derivative i as described, for example, in Grega, K. C; et al. in J. Org. Chem. 1995, 60, 5255-5261 and
Hutehinson, D. K.; et al. PCT hit. Appl. WO 9709328. It will be obvious to one skilled in the art that variations in the silyl protecting group are possible. Alternatively, the aniline a can be converted to the protected dimethylpyrrole derivative j. Treatment of either i or j with n-BuLi in THF affords an intermediate lithiated phenyl ring which can be added to tetrahydrothiopyran-4-one (k) to give an intermediate adduct. The silyl protecting group can be removed by stirring with methanolic potassium carbonate or the like. Subsequent reaction with an appropriate alkyl chloroformate then affords the carbamate 1. In the case of the dimethylpyrrole- protected intermediate adduct, deprotection can be achieved by treatment with hydroxylamine in aqueous ethanol. Again, subsequent reaction with an appropriate alkyl chloroformate then affords the carbamate 1. Closely related chemistry pertinent to this coupling chemistry can be found in Herrinton, P. M.; et al. Org. Process Res. Develop. 2001, 5, 80-83 and in Poel, T. J.; et al. PCT Int. Appl. WO 9929688. Dehydration of the alcohol moiety of 1, as described in Gage, J. R.; et al. Tetrahedron Lett. 2000, 41, 4301-4305, or variations thereof, then provides the unsaturated dihydropyran intermediate. Further transformation of intermediate m to advanced tetrahydropyran intermediate o can be conducted as described in the above references. Conversion of o to the desired tetrahydropyran examples p is accomplished by methods described for compounds in Schemes I and II and also as outlined in the above references. The unsaturated dihydropyran intermediate m can also be elaborated to the dihydropyran oxazolidinone derivative q The double bond of q can
be reduced as described in the above references to provide the corresponding tetrahydropyran oxazolidinones p.
SCHEME IV
EXAMPLE 1 Preparation of (S)-N-[[3-[2,3,5-trifluoro-4-(thiomorpholin-4- yl)phenyl]-2-oxo-5-oxazolidinonyl]methyl]acetamide.
Step 1 : Preparation of 4-(2,3 ,6-trifluoro-4-nitrophenyl)thiomorpholine
A solution of 2,3,4,5-tetrafluoronitrobenzene (61.0 mmol) in DMSO (150 mL) is treated with N,N-diisopropylethylamine (61.0 mmol) and then thiomorpholine (61.0 mmol) in acetonitrile (40 mL) is added dropwise at room temperature. After stirring 8 hours at room temperature the mixture is diluted with H O and extracted with EtOAc. The combined organic extracts are washed with H O, brine, dried over
Mg SO
4, filtered and concentrated under reduced pressure. The residue is triturated with Et
2O. The solid is collected by filtration and chromatographed over silica gel (Biotage 40M column), eluting with 1:1 dichlormethane /10% EtOAc/hexane. Appropriate fractions are combined and concentrated under reduced pressure to give the title compound.
Step 2: Preparation of 4-[4-(benzyloxycarbonyl)amino-2,3,6-trifluorophenyl] thiomorpholine
4-(2,3,6-trifluoro-4-nitrophenyl)thiomo holine (72.8 mmol) is suspended in
EtOH (250 mL) and treated with stannous chloride dihydrate (291.2 mmol). The mixture is then heated to reflux temperature. After 1 hour TLC analysis (10% EtOAc / hexanes) revealed the reduction to be complete. The reaction mixture is cooled to room temperature and poured into ice / H2O. The mixture is made basic by the addition of 2M sodium hydroxide and then stirred for 8 hour at room temperature and then overnight at room temperature. Added H2O and extracted with EtOAc. Combined organic extracts are washed with H2O, brine, dried over Mg2SO4, filtered and concentrated under reduced pressure to give a crude product. Trituration with Et2O affordes the intermediate substituted aniline. The intermediate aniline is dissolved in THF (100 mL), cooled to 0 °C, and treated 2M NaOH (20 mL, 35.0 mmol) and then benzyloxychloroformate (3.58 g, 3.0 mL, 21.0 mmol). The cooling bath is then removed and the mixture allowed to reach ambient temperature over an 8 hour period. After additional time at room temperature the reaction is judged to be essentially complete by TLC (50% EtOAc / hexanes). The mixture is poured into H2O and extracted with EtOAc. Combined organic extracts are washed with H2O, brine, dried over Mg2SO4, filtered, concentrated under reduced pressure and triturated with Et2O to give the title compound.
Step 3: Preparation of tert-butyl (5)-N-[[3-[2,3,5-trifluoro-4-(thiomorpholin-4- yl)phenyl]-2-oxo-5-oxazolidinonyl] methyljcarbamate

The starting material, 4-[4-(benzyloxycarbonyl)amino-2,3,6-trifluorophenyl] thiomorpholine (10.95 mmol), is dissolved in DMF (20 mL) and the mixture cooled to 0 °C. Lithium tert-butoxide (33.0 mL of a 1.0M solution in hexane, 32.85 mmol) is added dropwise via an addition funnel. When the addition is complete, tert-butyl (S)- N-(3-chloro-2-hydroxyprop-l-yl)carbamate (21.9 mmol) is added in one portion. The cooling bath is then removed and the mixture allowed to warm to ambient temperature over 8 hour and then left overnight at ambient temperature. TLC analysis (1:1 EtOAc / hexane) revealed the reaction to be nearly complete. The reaction mixture is quenched with 0. IN aqueous HCl. An aqueous workup with EtOAc afforded a crude product which is chromatographed on a Biotage silica gel column (40M), eluting with 1:1 EtOAc / hexane. Appropriate fractions are combined and concentrated under reduced pressure. Trituration with Et O afforded a solid which is isolated and dried under vacuum to give 1.77 g (37%) of the title compound as a white solid. Step 4: Preparation of (5)-N-[[3-[2,3,5-trifluoro-4-(thiomorpholin-4- yl)phenyl]-2-oxo-5-oxazolidinonyl]methyl]acetamide
rert-Butyl (5)-N-[[3-[2,3,5-trifluoro-4-(thiomorpholin-4-yl)phenyl]-2-oxo-5- oxazolidinonyl] methyl] carbamate (4.10 mmol) is suspended in dichloromethane, cooled to 0 °C and treated with trifluoroacetic acid (3 mL). After 2 hours, TLC analysis (10% MeOH / dichloromethane) reveals the reaction to be incomplete so the cooling bath is removed and the mixture is allowed to warm to ambient temperature overnight. The mixture is then concentrated under reduced pressure and the residue chromatographed over silica gel (Biotage column), eluting with 3 to 5 % MeOH/ dichloromethane/ammonia). Appropriate fractions are combined and concentrated under reduced pressure. Trituration with Et2O afforded, after drying, 1.16 g (85%) of
the intermediate 5-(aminomethyl)oxazolidinone as a white solid. The following characteristics are noted: HRMS calc'd. for C
14H
17F
3N
3O
3 (M+H): 332.1222; Found: 332.1216. Analytical calc'd. for C
14H
16F
3N
3O
3: C, 50.76; H, 4.87; N, 12.68; Found: C, 50.13; H, 4.83; N, 12.45. The intermediate amine (1.42 mmol) is dissolved in dichloromethane (10 mL), cooled to 0 °C and treated with pyridine (0.22 g, 0.23 mL, 2.84 mmol) and acetic anhydride (0.16 g, 0.15 mL, 1.56 mmol). The cooling bath is removed and the reaction mixture warmed to room temperature for 2 hours. The mixture is quenched with 1M aqueous HCl. The mixture is extracted with EtOAc. The combined organic extracts are washed with H
2O, brine, dried over Mg
2SO
4, filtered and concentrated under reduced pressure to give a crude product.
Chromatography over silica gel (Biotage column), eluting with 1 to 2% MeOH / dichloromethane afforded, after combination of appropriate fractions and concentration under reduced pressure, the purified material. Trituration with Et2O provided, after drying under reduced pressure, 0.422 g (80%) of the title compound as a white solid. 1H NMR (300 MHz, DMSO- 6) δ 8.25 (m, 1 H), 7.33 (ddd, 7= 3, 6, 12 Hz, 1 H), 4.76 (m, 1 H), 4.06 (1, 7= 9 Hz, t H), 3.70 (dd, 7= 6, 9 Hz, 1 H), 3.42 (m, 4 H), 2.70 (m, 4 H), 1.85 (s, 3 H); 13C NMR (75 MHz, CDCI3J δ 169.9, 154.5, 140.7, 133.3, 128.2 (m), 121.2 (m), 108.3 (d, 7 = 24 Hz), 72.9, 52.8, 48.6, 41.1, 27.4, 22.3.
EXAMPLE 2 Preparation of N-({ (5S)-2-oxo-3-[2,3,5-trifluoro-4-(l- oxidothiomorpholin-4-yl)phenyl]- 1 ,3-oxazoli-din-5-yl }methyl)acetamide.
(5)-N-[[3-[2,3,5-trifluoro-4-(thiomorpholin-4-yl)phenyl]-2-oxo-5- oxazolidinonyl]methyl] acetamide (1.35 mmol) is suspended in 40-mL of 1:1 MeOH:H2O and then 0.35 g of NaIO4 (1.62 mmol) is added in one portion. The reaction is monitored by TLC until complete (ca. 3 hours). When determined to be complete, the reaction is filtered through a short plug of Celite, and the filtrate is
concentrated in vacuo. The crude residue is subjected to Biotage chromatography (adsorbed onto SiO2, 40M) with 4% MeOH/CH2Cl2 to afford the desired sulfoxide compound (in 69% yield). 1H NMR (400 MHz, DMSO- 6) δ 8.26 (m, 1 H), 7.37 (ddd, 7 =2, 8, 16 Hz, 1 H), 4.77 (m, 1 H), 4.07 (t, 7 = 8 Hz, 1 H), 3.80 (m, 2 H), 3.71 (dd, 7 = 8, 9 Hz, 1 H), 3.42 (m, 2 H), 3.20 (m, 2 H), 2.99 (m, 2 H), 2.83 (m, 2 H), 1.85 (s, 3 H); 13C NMR (75MHz, CDC13) δ 169.9, 154.5, 152.5 (ddd, 7= 246, 6, 2 Hz), 146.4 (ddd, 7 = 247, 14, 9 Hz), 142.5 (ddd, 7 = 249, 14, 4 Hz), 127.4 (dd, 7 = 16, 11 Hz), 121.4, 108.3 (d, 7= 27 Hz), 72.9, 48.6, 45.5, 41.9, 41.1, 22.3; [α]25 D = -12 (c 0.97, DMSO).
EXAMPLE 3 Preparation of N-({(5S)-2-oxo-3-[2,3,5-trifluoro-4-(l,l- dioxidothiomorpholin-4-yl)phenyl]-l,3-oxazolidin-5-yl}methyl)acetaaade.
(5')-N-[[3-[2,3,5-trifluoro-4-(thiomorpholin-4-yl)ρhenyl]-2-oxo-5- oxazolidinonyl]methyl]acetamide (1.35 mmol) is suspended in (1.33 mmol) is suspended in 10 mL of 1:1 acetone: water, and then OxoneTM (1.06 g, 1.73 mmol) is added. The reaction progress is monitored by TLC and determined to be complete after 2 hours at room temperature. The solid precipitate is filtered, and then dissolved in CH2C12/MeOH and adsorbed onto SiO2. Biotage chromatography with 4%
MeOH/CH2C12 afforded the desired sulfone compound in 76% yield. 1H NMR (400 MHz, DMSO-J6) δ8.26 (m, 1 H), 7.38 (ddd, 7= 1, 4, 12 Hz, 1 H), 4.77 (m, 1 H), 4.07 (t, 7= 8 Hz, 1 H), 3.71(dd, 7 = 4, 8 Hz, 1 H), 3.58 (m, 4 H), 3.48-3.36 (m, 2 H), 3.24 (m, 4 H), 1.85 (s, 3 H); 13C NMR (75 MHz, CDCla) 6170.0, 154.5, 152.3 (dd, 7 = 241, 5 Hz), 146.3 (ddd, 7 = 247, 13, 9 Hz), 142.4 (ddd, 7 = 248, 14, 4 Hz), 126.5 (dd, 7= 11, 16 Hz), 121.6, 108.2 (d, 7= 28 Hz), 72.9, 51.75, 49.4, 41.1, 22.3.
EXAMPLE 4 Preparation of (S)-N-[[3-[2,3,5-trifluoro-4-[4-
(hydroxyacetyl)piperazin-l-yl]phenyl]-2-oxo-5-oxazolidinonyl]methyl]acetamide.
Step 1: Preparation of l-(benzyloxycarbonyl)-4-(2,3,6-trifluoro-4- nitrophenyl)piperazine
A solution of 2,3,4,5-tetrafluoronitrobenzene (10.0 g, 51.2 mmol) in DMSO (200 mL) is treated with N,N-diisopropylethylamine (6.95 g, 8.8 mL, 53.8 mmol) and then N-(benzyloxycarbonyl)piperazine (Aldrich, 11.8 g, 53.8 mmol) is added over 30 min at room temperature. The mixture is stirred overnight at ambient temperature. The mixture is diluted with H O and the extracted with EtOAc. The combined organic extracts are washed with H
2O, brine, dried over Mg
2SO
4, filtered and concentrated under reduced pressure. The residue is triturated with Et O / hexanes. The solid is collected by filtration and dried under vacuum to give 18.70 g (92%) of the title compound as a light yellow solid. HRMS calc'd. for C
18H
16F
3Ν
3O
4: 396.1171; Found: 396.1190. Analytical calc'd. for C
18H
16F
3N
3O
4: C, 54.69; H, 4.08; N, 10.63; Found: C, 54.56; H, 4.12; N, 10.62. Step 2: Preparation of l-(benzyloxycarbonyl)-4-[4-(benzyloxycarbonyl)amino-
2,3 ,6-trifluorophenyl]piperazine
l-(benzyloxycarbonyl)-4-(2,3,6-trifluoro-4-nitrophenyl)piperazine (18.70 g, 47.3 mmol) is suspended in EtOH (300 mL) and treated with stannous chloride dihydrate (42.6 g, 189.2 mmol). The mixture is then heated to reflux temperature. After 2 hours TLC analysis (30% EtOAc / hexanes) revealed the reduction to be complete. The reaction mixture is cooled to rt and poured into ice / H
2O. The mixture is made basic by the addition of 2M sodium hydroxide and then stirred for 3 at rt. The mixture is poured into more ice / H
2O, made basic (pH 10) with 2M sodium hydroxide and stirred another 10 h. Added H
2O and extracted with EtOAc. Combined organic extracts are washed with H O, brine, dried over Mg
2SO
4, filtered
and concentrated under reduced pressure to give the crude product. Trituration with Et
2O afforded a cleaner material (10.27 g) with: HRMS calc'd. for C
18H
18F
3N
3O
2: 366.1429; Found: 366.1414. The intermediate aniline (10.0 g, 27.4 mmol) wa sdissolved in THF, cooled to 0 °C, and treated 2M NaOH (50 mL) and then benzyloxychloroformate (18.7 g, 16.0 mL, 109.5 mmol). The cooling bath is then removed and the mixture allowed to reach ambient temperature over an 8 hours period. After an additional 16 hours at room temperature the reaction is judged to be essentially complete by TLC (30% EtOAc / hexanes). The mixture is poured into H
2O and extracted with EtOAc. Combined organic extracts are washed with H
2O, brine, dried over Mg
2SO
4, filtered and concentrated under reduced pressure to give 11.3 g (83%) of the crude title product. HRMS calc'd. for C
26H
24F
3N
3O
4: 500.1797; Found: 500.1797.
Step 3: Preparation of 7/ert-Butyl (S)-N-[[3-[2,3,5-trifluoro-4-[4-
(benzyloxycarbonyl)piperazin-l-yl]phenyl]-2-oxo-5-oxazolidinonyl]methyl]carbamate
The starting material, l-(benzyloxycarbonyl)-4-[4-(benzyloxycarbonyl)amino- 2,3,6-trifluorophenyl]piperazine (11.16 g, 22.3 mmol), is dissolved in DMF (40 mL) and the mixture cooled to 0 °C. Lithium tert-butoxide (67.0 mL of a 1.0M solution in hexane, 66.9 mmol) is added dropwise via an addition funnel. When the addition is complete, tert-butyl (ιS)-N-(3-chloro-2-hydroxyprop-l-yl)carbamate (9.35 g, 44.6 mmol) is added in one portion. The cooling bath is then removed and the mixture allowed to warm to ambient temperature over 8 hours and then left at rt overnight. The reaction mixture is quenched with 0.1N aqueous HCl. An aqueous workup with EtOAc afforded a crude product which is chromatographed on a Biotage silica gel column, eluting with 1:1 EtOAc / hexane. Appropriate fractions are combined and concentrated under reduced pressure. Trituration with Et
2O afforded a solid which is isolated and dried under vacuum to give 6.44 g (51%) of the title compound (also see International Patent Publication No. WO 02/085849 A3) HRMS calc'd. for
C
27H
31F
3N
4O
6: 565.2274; Found: 565.2255. Analytical calc'd. for C
27H
31F
3N
4O
6: C,
57.44; H, 5.53; N, 9.92; Found: C, 57.45; H, 5.54; N, 9.93.
Step 4: (5 -N-[[3-[2,3,5-trifluoro-4-[4-(benzyloxycarbonyl)piperazin-l- yl]phenyl]-2-oxo-5-oxazolidinonyl]methyl]acetamide
Tert-Butyl (>S)-N-[[3-[2,3,5-trifluoro-4-[4-(benzyloxycarbonyl)piperazin-l- yl]phenyl]-2-oxo-5-oxazolidinonyl]methyl]carbamate (1.00 g, 1.77 mmol) is suspended in MeOH and cooled to 0 °C. Then HCl in dioxane (1.5 mL of a 1.4M solution) is added and reaction progress monitored by TLC. After 8 hours additional HCl in dioxane (3 mL) is added and the mixture stirred at room temperature for an additional 16 hours. The mixture is concentrated under reduced pressure and triturated with Et
2O to give the crude hydrochloride salt of the 5-aminomethyl oxazolidinone. This crude product is suspended in dichloromethane, cooled to 0 °C and treated with pyridine (2 mL) and acetic anhydride (1 mL). The reaction is worked up after 1 hour by diluting with H O and then extracting with EtOAc. The combined organic extracts are washed with H
2O, brine, dried over Mg
2SO , filtered and concentrated under reduced pressure to give a crude product. Trituration with Et O provided, after drying under reduced pressure, 0.712 g (79%) of the title compound as a white solid. HRMS calc'd. for C
24H
25F
3N
4O
5: 507.1855; Found: 507.1857.
Step 5: Preparation of (1S)-N-[[3-[2,3,5-trifluoro-4-(4-piperazin-l-yl)phenyl]-2- oxo-5-oxazolidinonyl]methyl]acetamide
(S)-N-[[3-[2,3,5-trifluoro-4-[4-(benzyloxycarbonyl)piperazin-l-yl]phenyl]-2-oxo-5- oxazolidinonyl]methyl]acetamide (0.580 g, 1.15 mmol) is dissolved in 5:1 THF /
MeOH and placed under a nitrogen atmosphere. 10% Palladium on carbon (100 mg) is added and the reaction mixture stirred under H
2 (balloon) at rt. Reaction progress is monitored by TLC. After 12 hours the reaction mixture is filtered through Celite and concentrated under reduced pressure. The crude product is chromatographed over silica gel (Biotage column), eluting with 3% MeOH / dichloromethane. Appropriate fractions are combined and concentrated under reduced pressure. Trituration of the residue afforded 0.337 g (79%) of the title compound as a white solid. HRMS calc'd. for Cι
6H
19F
3N
4O
3: 373.1487; Found: 373.1494. Step 6: Preparation of (5)-N-[[3-[2,3,5-trifluoro-4-[4- (hydroxyacetyl)piρerazin-l-yl]ρhenyl]-2-oxo-5-oxazolidinonyl]methyl]acetamide
(5)-N-[[3-[2,3,5-trifluoro-4-(4-piperazin-l-yl)phenyl]-2-oxo-5- oxazolidinonyljmethyl] acetamide (0.243 g, 0.65 mmol) in dichloromethane (10 mL) is cooled to 0 °C and treated first with pyridine (0.13 mL, 1.56 mmol) and then acetoxyacetyl chloride (0.08 mL, 0.78 mmol). After 1 hour the reaction mixture is quenched with H
2O and then extracted with dichloromethane. The combined organic extracts are washed with H
2O, 1M aqueous HCl, brine, dried over MG SO
4, filtered and concentrated under reduced pressure to give the crude product as a white foam. Trituration with Et
2O afforded 0.1508 g (49%) of the desired (5)-N-[[3-[2,3,5- trifluoro-4-[4-(acetoxyacetyl)piperazin-l-yl]phenyl]-2-oxo-5- oxazolidinonyl]methyl] acetamide intermediate which is used directly in the next step. The material is suspended in 3: 1 MeOH / H
2O (10 mL), cooled to 0 °C and treated with lithium hydroxide (0.03 g, 0.64 mmol). After 4 hours the reaction mixture is quenched with IN aqueous HCl (10 mL). The methanol is removed by rotary evaporation under reduced pressure and the aqueous layer extracted with dichloromethane. The combined organic extracts are washed with H O, brine, dried over Mg
2SO , filtered and concentrated under reduced pressure. The crude product is chromatographed over silica gel (Biotage column), eluting with 3% MeOH / dichloromethane, to give, after concentration of appropriate fractions, trituration with Et
2O and drying under reduced pressure, 0.048 g (35%) of the title compound as a
white solid. HRMS calc'd. for C
18H
2ιF
3N
4O
5: 431.1542; Found: 431.1540. Analytical calc'd. for C
18H
21F
3N
4O
5: C, 50.23; H, 4.92; N, 13.02; Found: C, 49.85; H, 5.00; N, 12.84. Following the general procedure of the above Example, and making non- critical variations but substituting a different either for Et O in of Step 4, the corresponding piperazinyl trifluorphenyl oxazolidinones whereing R
1 is C
-6 alkyl can be obtained.
EXAMPLE 5 Preparation of (S)-N-[[3-[2,3,5-trifluoro-4-(morpholin-4- yl)phenyl]-2-oxo-5-oxazolidinonyl]methyl]acetamide
Step 1 Preparation of 4-(2,3,6-trifluoro-4-nitrophenyl)morpholine
A solution of 2,3,4,5-tetrafluoronitrobenzene (13.0 g, 61.0 mmol) in DMSO (150 mL) was treated with N,N-diisopropylethylamine (7.9 g, 9.96 mL, 61.0 mmol) and then morpholine (5.31 g, 5.32 mL, 61.0 mmol) in acetonitrile (40 mL) was added drop wise at room temperature. After stirring 8 h at rt the mixture was diluted with H
2O and extracted with EtOAc. The combined organic extracts were washed with H
2O, brine, dried over Mg
2SO
4, filtered and concentrated under reduced pressure. The residue was triturated with Et
2O. The solid was collected by filtration and chromatographed over silica gel (Biotage 40M column), eluting with 1:1 dichloromethane / 10% EtOAc/hexane. Appropriate fractions were combined and concentrated under reduced pressure to give 15.2 g (95%) of the title compound as a yellow solid. Step 2 Preparation of 4-[4-(benzyloxycarbonyl)amino-2,3,6- trifluorophenyljmorpholine
4-(2,3,6-trifluoro-4-nitrophenyl)morpholine (19.1 g, 72.8 mmol) was suspended in EtOH (250 mL) and treated with stannous chloride dihydrate (65.7 g,
291.2 mmol). The mixture was then heated to reflux temperature. After 1 h TLC analysis (10% EtOAc / hexanes) revealed the reduction to be complete. The reaction mixture was cooled to rt and poured into ice / H
2O. The mixture was made basic by the addition of 2M sodium hydroxide and then stirred for 8 h at rt and then overnight at rt. Added H
2O and extracted with EtOAc. Combined organic extracts were washed with H
2O, brine, dried over Mg
2SO
4, filtered and concentrated under reduced pressure to give a crude product. Trituration with Et
2O afforded 16.55 g (98%) of the intermediate substituted aniline. The intermediate aniline (4.07 g, 17.5 mmol) was dissolved in THF (100 mL), cooled to 0 °C, and treated 2M NaOH (20 mL, 35.0 mmol) and then benzyloxychloroformate (3.58 g, 3.0 mL, 21.0 mmol). The cooling bath was then removed and the mixture allowed to reach ambient temperature over an 8 h period. After additional time at rt the reaction was judged to be essentially complete by TLC (50% EtOAc / hexanes). The mixture was poured into H
2O and extracted with EtOAc. Combined organic extracts were washed with H
2O, brine, dried over Mg
2SO , filtered, concentrated under reduced pressure and triturated with Et
2O to give the title product with the following characteristics: HRMS calc'd. for C
18Hι
8F
3N
2O
3 (M+H): 367.1269; Found: 367.1277. Analytical calc'd. for C
18H
17F
3N
2O
3: C, 59.01; H, 4.68; N, 7.65; Found: C, 58.99; H, 4.70; N, 7.68.
Step 3 Preparation of tert-Butyl (5)-N-[[3-[2,3,5-trifluoro-4-(morpholin-4- yl)phenyl]-2-oxo-5-oxazolidinonyl]methyl]carbamate
The starting material, 4-[4-(benzyloxycarbonyl)amino-2,3,6- trifluorophenyl]morpholine (4.01 g, 10.95 mmol), was dissolved in DMF (20 mL) and the mixture cooled to 0 °C. Lithium tert-butoxide (33.0 mL of a 1.0M solution in hexane, 32.85 mmol) was added drop wise via an addition funnel. When the addition was complete, tert-butyl (S N-(3-chloro-2-hydroxyprop-l-yl)carbamate (4.59 g, 21.9 mmol) was added in one portion. The cooling bath was then removed and the mixture
allowed to warm to ambient temperature over 8 h and then left overnight at ambient temperature. TLC analysis (1:1 EtOAc / hexane) revealed the reaction to be nearly complete. The reaction mixture was quenched with 0.1N aqueous HCl. An aqueous workup with EtOAc afforded a crude product which was chromatographed on a Biotage silica gel column (40M), eluting with 1 : 1 EtOAc / hexane. Appropriate fractions were combined and concentrated under reduced pressure. Trituration with Et
2O afforded a solid which was isolated and dried under vacuum to give 1.77 g (37%) of the title compound as a white solid with the following characteristics: HRMS calc'd. for C
19H
25F
3N
3O
5 (M+H): 432.1746; Found: 432.1747. Analytical calc'd. for C
19H
24F
3N3O
5: C, 52.90; H, 5.61; N, 9.74; Found: C, 52.96; H, 5.75; N, 9.68.
Step 4 Preparation of (S)-N-[[3-[2,3,5-trifluoro-4-(morpholin-4-yl)phenyl]-2-oxo-5- oxazolidinonyl]methyl]acetamide
rert-Butyl (5')-N-[[3-[2,3,5-trifluoro-4-(morpholin-4-yl)phenyl]-2-oxo-5- oxazolidinonyl]methyl]carbamate (1.77 g, 4.10 mmol) was suspended in dichloromethane, cooled to 0 °C and treated with trifluoroacetic acid (3 mL). After 2 h, TLC analysis (10% MeOH / dichloromethane) revealed the reaction to be incomplete so the cooling bath was removed and the mixture allowed to warm to ambient temperature overnight. The mixture was then concentrated under reduced pressure and the residue chromatographed over silica gel (Biotage column), eluting with 3 to 5 % MeOH / (dichloromethane/ammonia). Appropriate fractions were combined and concentrated under reduced pressure. Trituration with Et2O afforded, after drying, 1.16 g (85%) of the intermediate 5-(aminomethyl)oxazolidinone as a white solid. The following characteristics were noted: HRMS calc'd. for C
14H
17F
3N
3O
3 (M+H): 332.1222; Found: 332.1216. Analytical calc'd. for C
14H
16F
3N
3O
3: C, 50.76; H, 4.87; N, 12.68; Found: C, 50.13; H, 4.83; N, 12.45. The intermediate amine (0.470 g, 1.42 mmol) was dissolved in dichloromethane (10 mL), cooled to 0 °C and treated with pyridine (0.22 g, 0.23 mL, 2.84 mmol) and acetic
anhydride (0.16 g, 0.15 mL, 1.56 mmol). The cooling bath was removed and the reaction mixture warmed to rt for 2 h. The mixture was quenched with 1M aqueous HCl. The mixture was extracted with EtOAc. The combined organic extracts were washed with H
2O, brine, dried over Mg
2SO , filtered and concentrated under reduced pressure to give a crude product. Chromatography over silica gel (Biotage column), eluting with 1 to 2% MeOH / dichloromethane afforded, after combination of appropriate fractions and concentration under reduced pressure, the purified material. Trituration with Et
2O provided, after drying under reduced pressure, 0.422 g (80%) of the title compound as a white solid with the following characteristics: HRMS calc'd. for C
18H
19F
3N
3O
4 (M+H): 374.1328; Found: 374.1320. Analytical calc'd. for C
18H
18F
3N
3O
4: C, 51.48; H, 4.86; N, 11.26; Found: C, 51.31; H, 4.83; N, 11.18. Specific rotation: [α]
D -12°. The method of Example 5, with the substitution of different acid anhydrides in step d) may be used to prepare other amides of formula I.
Activity Data The in vitro activity of the compound of this invention can be assessed by standard testing procedures such as the determination of minimum inhibitory concentration (MIC) by agar dilution as described in "Approved Standard. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically", 3rd. ed., published 1993 by the National Committee for Clinical Laboratory Standards, Villanova, Pennsylvania, USA. The activity of compounds of this invention against Staphylococcus aureus is shown in Table 1. Oxazolidinones with weak or no ability to inhibit MAO-A have the potential to minimize or eliminate potential drug-drug interactions. The compound of the present invention is tested for their MAO inhibitory activity by using the procedures below. The enzyme assay for human MAO-A relies on the formation of a colored reaction product by the enzyme. This product is detected by a spectrophotometer at 421 nm. The chromogenic substrate is l-methyl-4-(l-methyl-2-pyrryl)-l,2,3,6- tetrahydropyridine. Membrane bound, human placental MAO-A is solubilized and purified as described and used as a concentrated solution (5 nmoles per ml). See:
Flaherty P, Castagnoli K, Wang Y-X, Castagnoli Jr. N., JMed Chem. Vol. 39, p. 4756, (1996). Stock Solutions- Sodium phosphate is prepared as a 50 mM stock solution, pH 7.3 at 37°C. Stock solutions (50 mM) of the test compounds are prepared in DMSO. Serial dilutions of the 50 mM stocks are made in DMSO to form additional stock solutions ranging from 20 mM to 0.3125 mM. These stocks are then frozen until needed. The stocks are diluted 1/100 into the final enzyme assay volume at the time of assay. A 10 mM stock solution of the chromogenic substrate is prepared in the 50 mM phosphate buffer, aliquoted and then frozen until time of use. Enzyme Assay- Initial velocity assays are run in a SPECTRAmax 250 microplate spectrophotometer (Molecular Devices Corp., Sunnyvale, CA.). The final composition of the assay solution is 0.05 M sodium phosphate, pH 7.3, 80 mM substrate, inhibitor concentrations ranging up to 500 mM, 1% DMSO, and sufficient enzyme to produce an absorbency change at 421 nm of 0.0015-0.002/ min. The reactions are run at 37 °C. The reaction is followed by recording the increase in absorbency at 421 nm. Inhibitors are pre-incubated with the MAO-A in the reaction mixture for 15 min. prior to starting the reaction. Ki values are determined from the initial velocity data using the following equation. See Segel, I.H., Enzyme Kinetics. Vol. 957, p. 105, (1975). Wiley Merscience. NY., NY. %Inh. = 100*[I]/ (|TJ + Ki(l + [S]/Kni(s)). The results are also shown in Table 1.
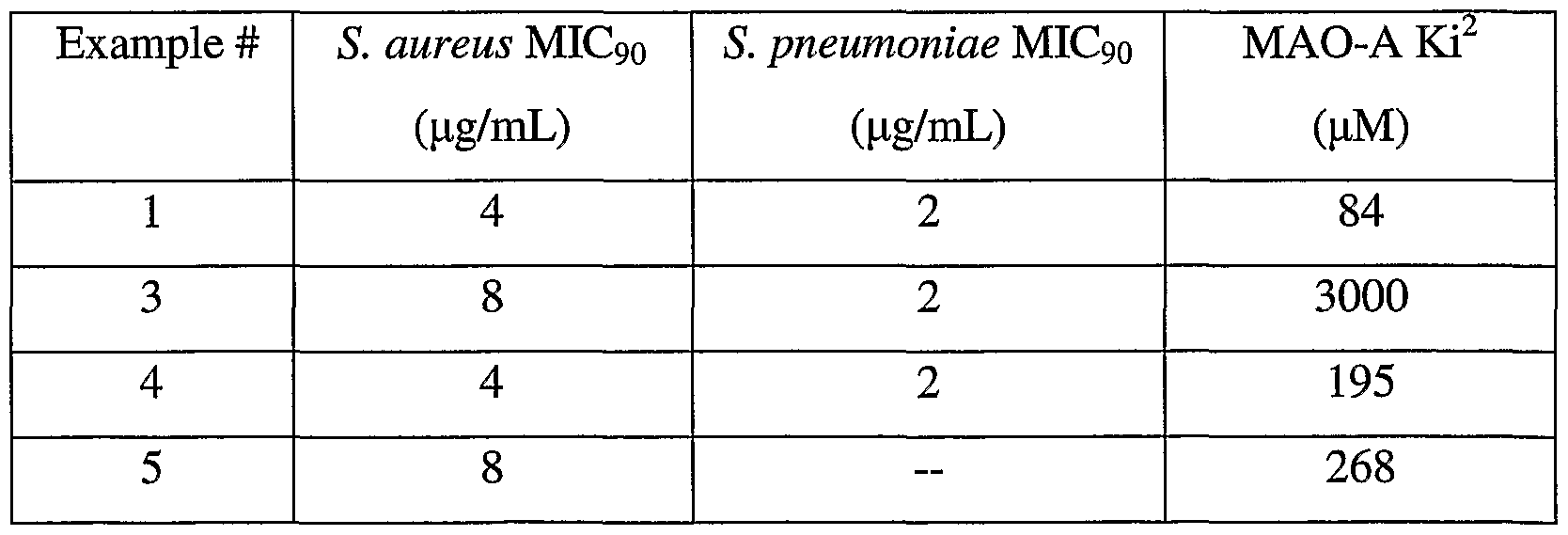
TABLE 1
^ICgo = minimum inhibitory concentration required to inhibit 90% of the isolates (lower number is better). 2Ki for human monoamine oxidase A (higher number is better).
The compound of the present invention, or the combination therapy has useful activity against a number of human and veterinary bacterial pathogens. Representative organisms include, but are not limited to, Staphylococcus aureus, Staphylococcus epidermidis, Enterococcus faecalis, Enterococcus faecium, Streptococcus pneumoniae, Streptococcus pyogenes, Chlamydophila pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Clostridium spp., Peptostreptococcus spp., Bacteroides spp, Listeria monocytogenes, Corynebacterium jeikeium, methicillin- resistant Staphylococcus aureus (MRS A), vancomycin-resistant Enterococci (VRE), glycopeptide-intermediate Staphylococcus aureus (GISA), and vancomycin- intermediate Staphylococcus aureus (VISA). It will be apparent to one skilled in the art that the described organisms are merely representative and that other bacteria are included within the spectrum of activity of the claimed compounds. Skin disease that may be treated using the compounds of the present invention include cutaneous anthrax, cellulites, necrotizing fasciitis, scalded skin syndrome, toxic epidermal necrolysis, furuncles, and erythrasma. Infectious diseases of the eye that may be treated using compounds of the present invention include orbital cellulites, dacrocystitis, blepharitis, hordeolum, bacterial conjunctivitis, and trachoma. Generally, an antibacterially effective amount of dosage of the compound of formula I of the present invention, either administered individually or in combination with other antibiotics, will be in the range of about 0.1 to about 400, more preferably about 1.0 to about 50 mg/kg of body weight/day. It is to be understood that the dosages of active component(s) may vary depending upon the requirements of each subject being treated and the severity of the bacterial infection. The desired dose may conveniently be presented in a single dose or as divided into multiple doses administered at appropriate intervals, for example, as two, three, four or more sub-doses per day. The sub-dose itself may be further divided, e.g., into a number of discrete loosely spaced administrations; such as multiple inhalations from an insufflator or by application of a plurality of drops into the eye. Also, it is to be understood that the initial dosage administered may be increased beyond the above upper level in order to rapidly achieve the desired plasma concentration. On the other hand, the initial dosage may be smaller than the optimum
and the daily dosage may be progressively increased during the course of treatment depending on the particular situation. Pharmaceutical compositions of the compound of formula I either individually or may be prepared by methods well known in the art, e.g., by means of conventional mixing, dissolving, granulation, dragee-making, levigating, emulsifying, encapsulating, entrapping, lyophilizing processes or spray drying. Pharmaceutical compositions for use in accordance with the present invention may be formulated in conventional manner using one or more physiologically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the active compounds into preparations that can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen. The compounds of formula I may be administered parenterally, orally, topically, transdermally, and rectally (e.g., as a suppository). Formulations for systemic administration may be in the form of aqueous solutions and suspensions, in addition to solid tablet and capsule formulations. The aqueous solutions and suspensions may be prepared from sterile powders or granules having one or more of the carriers or diluents mentioned for use in the formulations for oral administration. The compounds may be dissolved in water, polyethylene glycol, propylene glycol, ethanol, corn oil, cottonseed oil, peanut oil, sesame oil, benzyl alcohol, sodium chloride, and/or various buffers. Other adjuvants are well and widely known in the pharmaceutical art. For instance, the suspensions or solutions for systemic administration may include β-cyclodextrins, such as Captisol®, as a solubilizing agent. The compositions may, for example, be administered parenterally, e.g., intravascularly, intraperitoneally, subcutaneously, or intramuscularly. For parenteral administration, saline solution, dextrose solution, or water may be used as a suitable carrier. Formulations for parenteral administration may be in the form of aqueous or non-aqueous isotonic sterile injection solutions or suspensions. Pharmaceutical compositions for parenteral administration will generally contain a pharmaceutically acceptable amount of the compound or a soluble salt (acid addition salt or base salt) dissolved in a pharmaceutically acceptable liquid carrier such as, for example, water-for-injection and a buffer to provide a suitably buffered isotonic solution, for example, having a pH of about 3.5-6. Suitable buffering agents include, for example, trisodium orthophosphate, sodium bicarbonate, sodium citrate,
N-methylglucamine, L(+)-lysine and L(+)-arginine to name but a few representative buffering agents. The compound of this invention generally will be dissolved in the carrier in an amount sufficient to provide a pharmaceutically acceptable injectable concentration in the range of about 1 mg/mL to about 400 mg/mL of solution. The resulting liquid pharmaceutical composition will be administered so as to obtain the above-mentioned antibacterially effective amount of dosage. For systemic administration, the compounds can be formulated by combining the active compounds with pharmaceutically acceptable carriers well known in the art. Such carriers enable the compounds of the invention to be formulated as tablets, pills, lozenges, dragees, capsules, liquids, solutions, emulsions, gels, syrups, slurries, suspensions, and the like for oral ingestion by a patient. In addition to the compound of formula I, the pharmaceutical composition for therapeutic use may also comprise one or more non-toxic, pharmaceutically acceptable carrier materials or excipients. The term "carrier" material or "excipient" herein means any substance, not itself a therapeutic agent, used as a carrier and/or diluent and/or adjuvant, or vehicle for delivery of a therapeutic agent to a subject or added to a pharmaceutical composition to improve its handling or storage properties or to permit or facilitate formation of a dose unit of the composition into a discrete article such as a capsule or tablet suitable for oral administration. Excipients can include, by way of illustration and not limitation, diluents, disintegrants, binding agents, adhesives, wetting agents, polymers, lubricants, glidants, substances added to mask or counteract a disagreeable taste or odor, flavors, dyes, fragrances, and substances added to improve appearance of the composition. Acceptable excipients include stearic acid, magnesium stearate, magnesium oxide, sodium and calcium salts of phosphoric and sulfuric acids, magnesium carbonate, talc, gelatin, acacia gum, sodium alginate, pectin, dextrin, mannitol, sorbitol, lactose, sucrose, starches, gelatin, cellulosic materials, such as cellulose esters of alkanoic acids and cellulose alkyl esters, low melting wax, cocoa butter or powder, polymers such as polyvinyl- pyrrolidone, polyvinyl alcohol, and polyethylene glycols, and other pharmaceutical acceptable materials. The components pharmaceutical composition can be encapsulated or tableted for convenient administration. For oral administration, the pharmaceutical composition may be in the form of, for example, a tablet, capsule, suspension, or liquid. If desired, other active
ingredients may be included in the composition. The suspension or liquid may include other additives such as β-cyclodextrins, such as Captisol®, which may act as a solubilizing agent. Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses. Pharmaceutical compositions, which can be used orally, include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. The push-fit capsules can contain the active ingredients in admixture with a filler such as lactose, a binder such as starch, and/or a lubricant such as talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, liquid polyethylene glycols, cremophor, capmul, medium or long chain mono-, di- or triglycerides. Stabilizers may be added in these formulations, also. Liquid form compositions include solutions, suspensions and emulsions. For example, there may be provided solutions of the compounds of this invention dissolved in water and water-propylene glycol and water-polyethylene glycol systems, optionally containing suitable conventional coloring agents, flavoring agents, stabilizers and thickening agents. Alternatively, the compound of formula I may be in a powder form for constitution with a suitable vehicle, e.g., sterile, pyrogen-free water, before use. For suppository administration, the compounds may also be formulated by mixing the agent with a suitable non-irritating excipient which is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug. Such materials include cocoa butter, beeswax and other glycerides. As a topical treatment an effective amount of formula I is admixed in a pharmaceutically acceptable gel or cream vehicle that can be applied to the patient's skin at the area of treatment. Preparation of such creams and gels is well known in the
art and can include penetration enhancers, such as oils or alcohols, which increase or permit the compounds of formula I to penetrate the dermis to transdermal tissue. In some embodiments, the compound of formula I can be administrated by inhalation provided that the compounds pass into the blood stream. For example, pharmaceutical compositions containing the compound of formula I can be conveniently delivered through an aerosol spray in the form of solution, dry powder, or cream. The aerosol may use a pressurized pack or a nebulizer and a suitable propellant. In the case of a pressurized aerosol, the dosage unit may be controlled by providing a valve to deliver a metered amount. Capsules and cartridges of, for example, gelatin for use in an inhaler may be formulated containing a power base such as lactose or starch. Additionally, the compound of formula I may be delivered using a sustained- release system. Various sustained-release materials have been established and are well known by those skilled in the art. Sustained-release capsules may, depending on their chemical nature, release the compounds for 24 hours up to several days. Depending on the chemical nature and the biological stability of the therapeutic reagent, additional strategies for protein stabilization may be employed. The compound of formula I may also be delivered by controlled-release formulation as may be provided in a dispersion of active compound in hydroxypropyl- methyl cellulose, or other methods known to those skilled in the art.