WO2001040236A2 - Phosphorylation of terminal primary alcohol groups - Google Patents
Phosphorylation of terminal primary alcohol groups Download PDFInfo
- Publication number
- WO2001040236A2 WO2001040236A2 PCT/GB2000/004527 GB0004527W WO0140236A2 WO 2001040236 A2 WO2001040236 A2 WO 2001040236A2 GB 0004527 W GB0004527 W GB 0004527W WO 0140236 A2 WO0140236 A2 WO 0140236A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- isoxazol
- oxazolidin
- difluorophenyl
- yloxymethyl
- compound
- Prior art date
Links
- ZNXUQFFQYNELBR-QYQAVGFGSA-O N=C(/C=C\[OH2+])OCC(CN1c2cc(F)c(C3=CCN(Cc4ccccc4)CC3)c(F)c2)OC1=O Chemical compound N=C(/C=C\[OH2+])OCC(CN1c2cc(F)c(C3=CCN(Cc4ccccc4)CC3)c(F)c2)OC1=O ZNXUQFFQYNELBR-QYQAVGFGSA-O 0.000 description 1
- XADWCXJAUSYJFX-UHFFFAOYSA-N O=C(Nc1cc(F)c(C2=CCN(Cc3ccccc3)CC2)c(F)c1)OCc1ccccc1 Chemical compound O=C(Nc1cc(F)c(C2=CCN(Cc3ccccc3)CC2)c(F)c1)OCc1ccccc1 XADWCXJAUSYJFX-UHFFFAOYSA-N 0.000 description 1
- FZTYTRABDKQXQQ-LLVKDONJSA-N O=C1O[C@@H](COc2n[o]cc2)CN1c1cc(F)cc(F)c1 Chemical compound O=C1O[C@@H](COc2n[o]cc2)CN1c1cc(F)cc(F)c1 FZTYTRABDKQXQQ-LLVKDONJSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/091—Esters of phosphoric acids with hydroxyalkyl compounds with further substituents on alkyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6571—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and oxygen atoms as the only ring hetero atoms
- C07F9/6574—Esters of oxyacids of phosphorus
- C07F9/65742—Esters of oxyacids of phosphorus non-condensed with carbocyclic rings or heterocyclic rings or ring systems
Definitions
- the invention relates to chemical processes and chemical intermediates. More particularly, it relates to processes and intermediates which are useful in the selective formation of a primary mono-phosphoryl group in a terminal- 1 ,2-diol-propanoyl containing system, most particularly certain oxazolidinone anti-Gram positive bacterial agents containing such functionality.
- the invention also relates to processes for the manufacture of said intermediates and to processes for the manufacture of such oxazolidinone compounds utilising said intermediates.
- Co-pending International Patent Application No. GB99/01753 (WO 99/64417) describes a new class of antibacterial oxazolidinone compounds which are effective as anti- Gram positive bacterial agents, and certain processes for their preparation. Of the compounds disclosed, those of the formula (I) are included :
- HET is a C-linked 5-membered heteroaryl ring containing 2 to 4 heteroatoms independently selected from N, O and S, which ring is optionally substituted on an available carbon atom by 1 or 2 substituents independently selected from (l-4C)alkyl, amino, (l-4C)alkylamino, (1- 4C)alkoxy and halogen, and/or on an available nitrogen atom (provided that the ring is not thereby quaternised) by (l-4C)alkyl;
- R 2 and R 3 are independently hydrogen or fluoro;
- Rep is of the formula R 13p CO- (wherein R 13p is (l-lOC)alkyl substituted by two or more hydroxy groups; 2 of which are in a 1,2-diol orientation, ie. there is a terminal primary alcohol with an adjacent secondary alcohol), or pharmaceutically-acceptable salts, or in-vivo- hydrolysable esters thereof.
- R 13p is (l-lOC)alkyl substituted by two or more hydroxy groups; 2 of which are in a 1,2-diol orientation, ie. there is a terminal primary alcohol with an adjacent secondary alcohol
- pharmaceutically-acceptable salts or in-vivo- hydrolysable esters thereof.
- In-vivo hydro lysable esters include compounds of formula (I) and (I-l) in which any free hydroxy group independently forms a phosphoryl ester of the formula (PD3) :
- those of formula (I-l) are the pharmaceutically active anti-bacterial enantiomer.
- the pure enantiomer depicted in (I-l), or mixtures of the 5R and 5S enantiomers, for example a racemic mixture are included in GB99/01753. If a mixture of enantiomers is used, a larger amount (depending upon the ratio of the enantiomers) will be required to achieve the same effect as the same weight of the pharmaceutically active enantiomer.
- the enantiomer depicted below is the 5R enantiomer.
- the preparation described includes isolation of the primary mono-phosphoryl compound (Intermediate Example 15) from a mixture of compounds (including, for example, the bis- phosphoryl and cyclic phosphoryl compounds) by use of Medium Pressure Liquid Chromatography using ethyl acetate as the eluant.
- Other compounds of formula (I) and (I-l) above may be prepared using analagous chemistry.
- the detailed chemistry and reaction conditions employed is described in the accompanying non-limiting Examples, or is within the skill of the ordinary organic/medicinal chemist (see also WO 97/30995, the relevant process sections of which are inco ⁇ orated herein, for details on prepartion of certain intermediates).
- GB99/01753 discloses that for a compound of formula (I) and (I-l) containing a number of free hydroxy groups, those groups not being converted into a prodrug functionality may be protected (for example, using a t- butyl-dimethylsilyl group), and later deprotected. Also, enzymatic methods may be used to selectively phosphorylate or dephosphorylate alcohol functionalities.
- the prodrugs containing groups (PD3) may be prepared by reaction of a compound of formula (I) and (I- 1 ) containing suitable hydroxy group/s with a suitably protected phosphorylating agent (for example, containing a chloro or dialkylamino leaving group), followed by oxidation (if necessary) and deprotection.
- the "Existing Route" to the said compound although satisfactory, is not particularly suitable for the manufacture of large quantities of such products.
- Selectivity of reaction is important and can impact upon the yield of desired product obtained. Poor selectivity can also lead to the formation of undesired by-products which require removal.
- the "Existing Route” has potential difficulties associated with the selective formation, in good yield, of the primary mono-phosphoryl/secondary hydroxy moiety.
- Suitable leaving groups include halo (e.g. iodo, bromo, chloro), triflate or enol phosphate.
- the protected aniline used as the initial starting material may alternatively be protected as -N-[SiR 3 ] 2 where each R is independently a (l-4C)alkyl group, eg. -N-(SiMe 3 ) 2 .
- R is independently a (l-4C)alkyl group
- -N-(SiMe 3 ) 2 we have now discovered a number of further, convenient and useful, processes for the manufacture of said compound (and by analogy other compounds of formula (I) and (I- 1)), which reduces and/or converges the number of reaction stages and, reduces or removes the need for chromatographic purification of intermediates and/or final products.
- the invention also relates to the application of the chemistry described herein to any system requiring formation of a primary mono-phosphoryl group in a terminal- 1 ,2-diol-propanoyl containing system (such as, for example, 2,3-dihydroxypropanoyl and 3,4-dihydroxy-2-oxo- butyl).
- a terminal- 1 ,2-diol-propanoyl containing system such as, for example, 2,3-dihydroxypropanoyl and 3,4-dihydroxy-2-oxo- butyl.
- the invention relates to such 1 ,2-diol-propanoyl containing systems in a compound of formula (I) and (I-l), and most particularly to the said compound.
- the invention also relates to 1,2-diol-propanoyl containing systems in a compound of formula (I) and (I-l) wherein HET is a C-linked 6-membered heteroaryl ring containing 1 or 2 N, which ring is optionally substituted on any available C atom (provided that when the N atom is adjacent to the link, there is no substitution on any C atom that is adjacent to this N atom) by 1, 2 or 3 substituents independently selected from (l-4C)alkyl, amino, (l-4C)alkylamino, (l-4C)alkoxy, (l-4C)alkoxycarbonyl and halogen.
- Preferred 6- membered heteroaryl rings are pyridin-2-yl, pyridazin-3-yl or pyrazin-2-yl.
- the invention also relates to 1,2-diol-propanoyl containing systems in a compound of formula (I) and (I-l) wherein HET is a C-linked 5- or 6-membered heteroaryl ring as described herein, wherein the link to the oxazolidinone ring is via a thiomethyl (-CH 2 - S-) link rather than an oxymethyl (-CH 2 -O-) link (see claim 2 for compounds of formula(I-2)).
- the invention may also be used in 1 ,2-diol-propanoyl containing systems in a compound of formula (I) and (I-l) wherein HET is a C-linked 5- or 6-membered heteroaryl ring as described in WO 00/21960 (inco ⁇ orated herein by reference), wherein the link to the oxazolidinone ring is via an aminomethyl (-CH 2 -NH 2 -) link.
- HET is a C-linked 5- or 6-membered heteroaryl ring as described in WO 00/21960 (inco ⁇ orated herein by reference), wherein the link to the oxazolidinone ring is via an aminomethyl (-CH 2 -NH 2 -) link.
- protecting groups have been referred to.
- protecting groups see one of the many general texts on the subject, for example, 'Protective Groups in Organic Synthesis' by Theodora Green (publisher: John Wiley & Sons).
- Protecting groups may be used and removed by any convenient method as described in the literature or known to the skilled chemist as appropriate for the removal of the protecting group in question, such methods being chosen so as to effect removal of the protecting group with minimum disturbance of groups elsewhere in the molecule.
- the Schemes may be genericised to cover other analogous compounds of formula (I) and (I-l) and (1-2) mentioned herein (see claim 2 for (1-2)). Furthermore, the invention also relates to the application of the chemistry described herein to any system requiring formation of a primary mono-phosphoryl group (-OPO(OH) 2 ) in a terminal- 1 ,2-diol- propanoyl (HO-CH 2 CH(OH)-CO-) containing system.
- -OPO(OH) 2 a primary mono-phosphoryl group
- HO-CH 2 CH(OH)-CO- a terminal- 1 ,2-diol- propanoyl
- a particularly preferred process is that illustrated in Scheme 2B.
- the use of the hydroxy acid (2H) allows the formation of a protected primary 1 ,2-diol species (PgO- CH 2 CH(OH)-CO-, wherein Pg is a protecting group suitable for protecting alcohols and removable by acid, such as, for example, t-butyl, as in Compound (21) in the case of the said compound).
- Pg is a protecting group suitable for protecting alcohols and removable by acid, such as, for example, t-butyl, as in Compound (21) in the case of the said compound.
- the secondary phosphoryl compound which may be optionally protected, for example in the form of a phosphate ester, such as the t-butyl ester as in Compound (2J) in the case of the said compound).
- the secondary phosphoryl compound is in the form of a phosphate ester.
- a secondary phosphoryl compound suitable for use in the process may be obtained, for example, by standard phosphorylation chemistry, for example as described herein, using tert-butyl tetraethylphosphorodiimidite, or using phosphorous oxychloride or
- Scheme 3C is particularly preferred, and offers the advantageous use of the hydroxy acid (2H) to give the protected primary alcohol, and then selective secondary phosphorylation to give (3D); followed by coupling, and then by deprotection of the protected primary alcohol & predominant rearrangement of the secondary phosphoryl compound to the primary phosphoryl compound.
- Scheme 3C offers a particularly favourable route to the said compound, comprising comparatively few reaction stages in a convergent fashion.
- the coupling reaction of Schemes 3A to 3D may be carried out in the presence of a suitable base, for example, nBuLi at ca.
- a suitable inert solvent or diluent such as, for example, dimethylsulphoxide, 1 ,2-dimethoxyethane, tetrahydrofuran (THF), tetrahydropyran, diglyme or toluene.
- a preferred solvent is a mixture of THF and toluene.
- Schemes 3 A to 3D may also be achieved via Grignard chemistry, using, for example, an appropriate 4-bromo-phenyl compound in place of (IF).
- the starting materials for the reactions described herein may be obtained as described herein, or by analogy to such methods, or by standard procedures of organic chemistry.
- Intermediates (IE) and (IF) are preferred intermediates, especially (IF). 2.
- a leaving group other than Br could also be used.
- allyl alcohol could be used and the substitution performed in a reverse sense to that shown with a leaving group (e.g. chloro or mesylate) on (IB).
- Nosylate and mesylate may also be used in place of tosylate in the epoxide (IH- 1). Preferably nosylate is used.
- Example 1 shows retention of stereochemistry, i.e. (R)- and (S)-glycidyl nosylate give, respectively, (R)-, (S)- glycidyl ether product.
- the other epoxide isomer may be used as a starting material. If a racemate is obtained, chiral resolution/chromatography may be used to obtain the desired isomer.
- DIPEA is di-isopropyl-ethylamine.
- non-bulky protecting groups in place of t-Bu may be used in (2G) and (2H), for example, any (l-4C)alkyl group; any silyl group (for example trimethylsilyl); or a benzyl group (e.g. using acid catalysed removal, or a reductive removal using e.g. hydrogenation).
- (2F) may be converted at ambient temperature to, for example, the disodium salt by treatment with 2 mol.eq. sodium carbonate and working-up in acetone and then IMS. 5.
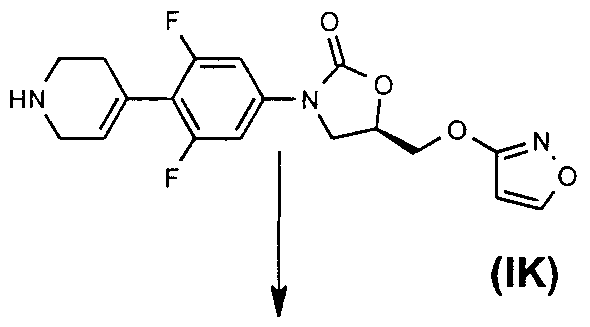
- (IK) may be prepared as shown in the Existing Route Scheme or as described in
- Example 4 hereinafter, in which, for example, Intermediate Example 2 may be prepared as follows :-
- a solution of 3,5-difluoroaniline in THF is chilled to -70°C.
- a solution of n-butyl lithium in toluene is added and chlorotrimethylsilane then added to complete the bis- trimethylsilyl protection of 3,5-difluoroaniline.
- a solution of n-butyl lithium in toluene is added to the chilled solution and a solution of l-benzyl-4-piperidone in toluene then added whilst maintaining the temperature.
- a solution of aqueous hydrochloric acid is added.
- the aqueous layer of the alcohol intermediate (Intermediate Example 1) is separated and heated to reflux while simultaneously distilling out tetrahydrofuran to complete the formation of Intermediate
- Example 2 The reaction is then diluted with water and butanol before adjusting the pH with aqueous ammonia at 40°C. The aqueous layer is separated and discarded. Cyclohexane is added to the organic phase to precipitate the product, which is then filtered off after cooling to ambient temperature, washed with a butanol/cyclohexane mixture, cyclohexane and dried under vacuum.
- Intermediate Example 4 may be prepared as described in Example 4 hereinafter, or using a solution of n-butyl lithium in toluene.
- (21) may be converted to Diol (2D) by deprotection, for example using acid conditions, such as HCl/dioxan.
- Hydrogen peroxide may be used in place of mCPBA in the conversion of (21) to
- reaction is performed in a suitable solvent, such as dioxan.
- R includes (l-4C)alkyl, for example, methyl, ethyl, propyl, iso-propyl, butyl, iso- butyl, tert-butyl; hydrogen and benzyl.
- (l-4C)alkyl includes both straight-chain and branched-chain alkyl groups.
- references to individual alkyl groups such as
- the lithiated compound may be transmetallated with, for example titanium chloride, titanium i-propoxide or cerium chloride at a tempertaure of about -30°C. Such transmetallation restricts enolisation of the piperidinone and so aids reaction at the desired centre.
- (3B) may be prepared from (2B) - see scheme 2A - using standard chemistry.
- (2H) may be prepared from O-t-Bu-serine (2G) using standard chemistry - see
- DMF is N,N-dimethylformamide
- DMA is
- Mobile phase A 10 mM ammonium acetate pH 4.5.
- Mobile phase B 10 mM ammonium acetate pH 4.5 in 90% acetonitrile.
- MIBK - DMF, acetone, toluene, MeCN, DME, NMP, THF, EtOAc, TBME, EtOH, MeOH.
- the following bases have also been used in the coupling reaction in place of caesium carbonate:- NaH, K 2 CO 3 , NaOH, NaOMe, NaOEt, KOMe, KOEt, KO'Bu, LDA, NEt 3 , NBu 3 , NPr 2 Et.
- 3-Hydroxyisoxazole may be prepared by cyclisation of CH ⁇ C-CO-NHOH (prepared from CH ⁇ C-CO-O-(l-4C)alkyl) as described in Chem.Pharm.Bull. Japan, 14, 92, (1966).
- 3-Hydroxyisoxazole may also be prepared as follows :- Hydroxylamine hydrochloride is neutralised with sodium hydroxide to liberate the free base. Ethyl propiolate in EtOH is then added dropwise maintaining the reaction temperature at 20-25°C and the reaction stirred before gradually warming to 50-55°C. Heating is continued at 50-55°C for 2.5h and the reaction is then acidified to pH ⁇ 3 with cone. HC1. On complete addition ca. 90% of the ethanol in the reaction is removed by distillation and the residue extracted with warm toluene. Toluene is removed by distillation to precipitate 3- hydroxyisoxazole, and the precipitation is completed by addition of cyclohexane. The resulting suspension is cooled and filtered prior to the material being dried in vacuo at ambient temperature.
- hydroxylamine hydrochloride is neutralised with sodium hydroxide.
- the hydroxylamine free base is reacted with a solution of ethyl propiolate in THF at 55 °C.
- the reaction mixture is cooled and acidified with hydrochloric acid, and the resulting solution extracted with butyronitrile, washed with dilute hydrochloric acid and the organic solution concentrated under reduced pressure to remove ethanol, THF and water.
- the solution may be used directly in a next stage.
- N-benzyloxycarbonyl-3,5-difluoroaniline intermediate is prepared by reaction of
- reaction mixture was cooled to 20-25 °C, and was washed with H 2 O (20 mL).
- the organic layer was separated, dried (MgSO 4 ) and was concentrated to give crude product that was purified by flash chromatography to give the desired product (IJ) (1.5g, 53 %).
- Mobile phase A 0.1% TFA in water.
- Mobile phase B 0.1% TFA in 90 % MeCN.
- Toluene - MIBK, THF, Toluene, TBME.
- HPLC showed 56% conversion to l-[4-(l-benzyl- l,2,3,6-tetrahydro-pyridin-4-yl)-3,5-difluoro-phenylamino]-3-(isoxazol-3-yloxy)-propan-2-ol (with reference to an external standard isolated in another reaction, which may be prepared by hydrolysis of Compound (IJ)).
- HPLC retention time (see below) 2.1 min.
- Mobile phase A 0.1% TFA in water.
- Mobile phase B 0.1% TFA in 90 % MeCN.
- THF was cooled to -70°C under nitrogen and 8.80ml of 1.6M nBuLi in hexanes (14.08mmol) added dropwise at the same temperature. After 20 minutes at the same temperature a solution of (R)-glycidyl butyrate (2.00g, 13.88mmol in 5ml THF) was added dropwise and the mixture stirred for 30 minutes at -70°C, and then stirred to ambient temperature overnight.
- Example 4 (2.6g, 6.5mmol), 3-hydroxyisoxazole (see Example 1; 0.60g, 7.06mmol), triphenylphosphine (1.96g, 7.48mmol) and diisopropylazodicarboxylate (1.44g, 7.13mmol) in THF (40ml) were reacted using the general method of Example 1.
- the resultant product was purified by flash chromatograpy (Merck 9385 silica, EtOAc / isohexane (3:2) eluant initially, then repeated using methyl tert-butylether eluant) to give the title product (2.6g, 86%) as a gum.
Abstract
Description




























Claims



Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001540991A JP2003515539A (en) | 1999-12-03 | 2000-11-28 | Chemical methods and intermediates |
| SK786-2002A SK7862002A3 (en) | 1999-12-03 | 2000-11-28 | Method of formation of a primary mono-phosphoryl group in a terminal 1,2-diol-propanoyl function group and intermediates used at this method |
| MXPA02005396A MXPA02005396A (en) | 1999-12-03 | 2000-11-28 | Chemical processes and intermediates. |
| AU17156/01A AU762241B2 (en) | 1999-12-03 | 2000-11-28 | Chemical processes and intermediates |
| BR0016087-3A BR0016087A (en) | 1999-12-03 | 2000-11-28 | Process for the formation of a primary mono-phosphoryl group in a terminal 1,2-diol-propanoyl functionality, processes for the preparation of 5- (het-x-methyl) -3- (4- (1-benzyl- 1,2,5,6-tetrahydropyrid-4-yl) - 3,5-difluorophenyl) oxazolidin-2-one, and 5-isoxazol-3-yloxymethyl-3- (4- (1-benzyl-1,2 , 5,6-tetrahydrop irid-4-yl) -3,5-difluorophenyl) oxazolidin-2-one, and, intermediate chemical compound |
| EP00979764A EP1237895A2 (en) | 1999-12-03 | 2000-11-28 | Phosphorylation of terminal primary alcohol groups |
| EEP200200282A EE200200282A (en) | 1999-12-03 | 2000-11-28 | Chemical methods and intermediates |
| HU0204052A HUP0204052A2 (en) | 1999-12-03 | 2000-11-28 | Phosphorylation of terminal primary alcohol groups |
| IL14964100A IL149641A0 (en) | 1999-12-03 | 2000-11-28 | Chemical processes and intermediates |
| CA002395052A CA2395052A1 (en) | 1999-12-03 | 2000-11-28 | Phosphorylation of terminal primary alcohol groups |
| KR1020027007072A KR20020058072A (en) | 1999-12-03 | 2000-11-28 | Chemical processes and intermediates |
| BG106728A BG106728A (en) | 1999-12-03 | 2002-05-20 | Chemical methods and intermediate compounds |
| IS6401A IS6401A (en) | 1999-12-03 | 2002-05-28 | Chemical methods and intermediates |
| NO20022605A NO20022605L (en) | 1999-12-03 | 2002-05-31 | Chemical processes and intermediates |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9928499.4A GB9928499D0 (en) | 1999-12-03 | 1999-12-03 | Chemical processes and intermediates |
| GB9928499.4 | 1999-12-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001040236A2 true WO2001040236A2 (en) | 2001-06-07 |
| WO2001040236A3 WO2001040236A3 (en) | 2002-05-10 |
Family
ID=10865576
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2000/004527 WO2001040236A2 (en) | 1999-12-03 | 2000-11-28 | Phosphorylation of terminal primary alcohol groups |
Country Status (22)
| Country | Link |
|---|---|
| EP (1) | EP1237895A2 (en) |
| JP (1) | JP2003515539A (en) |
| KR (1) | KR20020058072A (en) |
| CN (1) | CN1433421A (en) |
| AR (1) | AR026700A1 (en) |
| AU (1) | AU762241B2 (en) |
| BG (1) | BG106728A (en) |
| BR (1) | BR0016087A (en) |
| CA (1) | CA2395052A1 (en) |
| CO (1) | CO5271712A1 (en) |
| CZ (1) | CZ20021912A3 (en) |
| EE (1) | EE200200282A (en) |
| GB (1) | GB9928499D0 (en) |
| HU (1) | HUP0204052A2 (en) |
| IL (1) | IL149641A0 (en) |
| IS (1) | IS6401A (en) |
| MX (1) | MXPA02005396A (en) |
| NO (1) | NO20022605L (en) |
| PL (1) | PL364762A1 (en) |
| SK (1) | SK7862002A3 (en) |
| WO (1) | WO2001040236A2 (en) |
| ZA (1) | ZA200203876B (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002096916A1 (en) * | 2001-06-01 | 2002-12-05 | Astrazeneca Ab | Process for phosphorylation |
| US6919329B2 (en) | 2002-02-25 | 2005-07-19 | Pharmacia & Upjohn Company | N-Aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| US6969726B2 (en) | 2003-06-03 | 2005-11-29 | Rib X Pharmaceuticals Inc | Biaryl heterocyclic compounds and methods of making and using the same |
| US7022705B2 (en) | 2001-10-25 | 2006-04-04 | Astrazeneca Ab | Isoxazoline derivatives useful as antimicrobials |
| US7094900B2 (en) | 2002-08-12 | 2006-08-22 | Pharmacia & Upjohn Company Llc | N-Aryl-2-oxazolidinones and their derivatives |
| US7129259B2 (en) | 2003-12-17 | 2006-10-31 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141570B2 (en) | 2002-11-21 | 2006-11-28 | Pharmacia & Upjohn Company | N-aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| US7141588B2 (en) | 2002-02-25 | 2006-11-28 | Pfizer, Inc. | N-aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| US7199143B2 (en) | 2002-02-28 | 2007-04-03 | Astrazeneca Ab | Chemical compounds |
| US7304050B2 (en) | 2003-09-16 | 2007-12-04 | Pfizer Inc. | Antibacterial agents |
| US7335753B2 (en) | 2002-09-26 | 2008-02-26 | Rib-X Pharmaceuticals, Inc. | Bifunctional heterocyclic compounds and methods of making and using same |
| WO2008038092A2 (en) | 2006-09-25 | 2008-04-03 | Wockhardt Research Centre | Substituted piperidinophenyl oxazolidinones |
| US7396847B2 (en) | 2001-09-11 | 2008-07-08 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7473699B2 (en) | 2002-02-28 | 2009-01-06 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US8202843B2 (en) | 2004-02-27 | 2012-06-19 | Rib-X Pharmaceuticals, Inc. | Macrocyclic compounds and methods of making and using the same |
| US8324398B2 (en) | 2003-06-03 | 2012-12-04 | Rib-X Pharmaceuticals, Inc. | Process for the synthesis of biaryl oxazolidinones |
| US8399660B2 (en) | 2005-06-08 | 2013-03-19 | Rib-X Pharmaceuticals, Inc. | Process for the synthesis of triazoles |
| US9572809B2 (en) | 2012-07-18 | 2017-02-21 | Spero Trinem, Inc. | Combination therapy to treat Mycobacterium diseases |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999064417A2 (en) * | 1998-06-05 | 1999-12-16 | Astrazeneca Ab | Oxazolidinone derivatives, process for their preparation and pharmaceutical compositions containing them |
-
1999
- 1999-12-03 GB GBGB9928499.4A patent/GB9928499D0/en not_active Ceased
-
2000
- 2000-11-24 CO CO00090190A patent/CO5271712A1/en not_active Application Discontinuation
- 2000-11-28 CZ CZ20021912A patent/CZ20021912A3/en unknown
- 2000-11-28 HU HU0204052A patent/HUP0204052A2/en unknown
- 2000-11-28 JP JP2001540991A patent/JP2003515539A/en active Pending
- 2000-11-28 WO PCT/GB2000/004527 patent/WO2001040236A2/en not_active Application Discontinuation
- 2000-11-28 EE EEP200200282A patent/EE200200282A/en unknown
- 2000-11-28 BR BR0016087-3A patent/BR0016087A/en not_active Application Discontinuation
- 2000-11-28 AU AU17156/01A patent/AU762241B2/en not_active Ceased
- 2000-11-28 KR KR1020027007072A patent/KR20020058072A/en not_active Application Discontinuation
- 2000-11-28 IL IL14964100A patent/IL149641A0/en unknown
- 2000-11-28 EP EP00979764A patent/EP1237895A2/en not_active Withdrawn
- 2000-11-28 PL PL00364762A patent/PL364762A1/en unknown
- 2000-11-28 MX MXPA02005396A patent/MXPA02005396A/en unknown
- 2000-11-28 CN CN00818735A patent/CN1433421A/en active Pending
- 2000-11-28 CA CA002395052A patent/CA2395052A1/en not_active Abandoned
- 2000-11-28 SK SK786-2002A patent/SK7862002A3/en unknown
- 2000-12-01 AR ARP000106370A patent/AR026700A1/en unknown
-
2002
- 2002-05-15 ZA ZA200203876A patent/ZA200203876B/en unknown
- 2002-05-20 BG BG106728A patent/BG106728A/en unknown
- 2002-05-28 IS IS6401A patent/IS6401A/en unknown
- 2002-05-31 NO NO20022605A patent/NO20022605L/en not_active Application Discontinuation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999064417A2 (en) * | 1998-06-05 | 1999-12-16 | Astrazeneca Ab | Oxazolidinone derivatives, process for their preparation and pharmaceutical compositions containing them |
Non-Patent Citations (3)
| Title |
|---|
| BUCHWALD S.L.: "Stereochemical evidence for pseudorotation in the reaction of a phosphoric monoester" JOURNAL OF THE AMERICAN CHEMICAL SOCIETY., vol. 106, no. 17, 1984, XP002162618 AMERICAN CHEMICAL SOCIETY, WASHINGTON, DC., US ISSN: 0002-7863 * |
| KIS K.: "Biosynthesis of riboflavin." BIOCHEMISTRY., vol. 34, no. 9, 1995, pages 2883-2892, XP002162619 AMERICAN CHEMICAL SOCIETY. EASTON, PA., US ISSN: 0006-2960 * |
| MEYERHOF O.: "]ber die Isolierung der isomeren Phosphoglycerins{uren." BIOCHEMISCHE ZEITSCHRIFT., vol. 276, 1936, pages 239-253, XP000989557 SPRINGER, BERLIN., DE ISSN: 0366-0753 * |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002096916A1 (en) * | 2001-06-01 | 2002-12-05 | Astrazeneca Ab | Process for phosphorylation |
| US7396847B2 (en) | 2001-09-11 | 2008-07-08 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7022705B2 (en) | 2001-10-25 | 2006-04-04 | Astrazeneca Ab | Isoxazoline derivatives useful as antimicrobials |
| US6919329B2 (en) | 2002-02-25 | 2005-07-19 | Pharmacia & Upjohn Company | N-Aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| US7645781B2 (en) | 2002-02-25 | 2010-01-12 | Pfizer Inc | N-aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| US7141588B2 (en) | 2002-02-25 | 2006-11-28 | Pfizer, Inc. | N-aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| US7473699B2 (en) | 2002-02-28 | 2009-01-06 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7199143B2 (en) | 2002-02-28 | 2007-04-03 | Astrazeneca Ab | Chemical compounds |
| US7094900B2 (en) | 2002-08-12 | 2006-08-22 | Pharmacia & Upjohn Company Llc | N-Aryl-2-oxazolidinones and their derivatives |
| US7335753B2 (en) | 2002-09-26 | 2008-02-26 | Rib-X Pharmaceuticals, Inc. | Bifunctional heterocyclic compounds and methods of making and using same |
| US7141570B2 (en) | 2002-11-21 | 2006-11-28 | Pharmacia & Upjohn Company | N-aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| US7456206B2 (en) | 2003-06-03 | 2008-11-25 | Rib-X Pharmaceuticals, Inc. | Biaryl heterocyclic compounds and methods of making and using the same |
| US8324398B2 (en) | 2003-06-03 | 2012-12-04 | Rib-X Pharmaceuticals, Inc. | Process for the synthesis of biaryl oxazolidinones |
| US8895741B2 (en) | 2003-06-03 | 2014-11-25 | Melinta Therapeutics, Inc. | Process for the synthesis of biaryl oxazolidinones |
| US7148219B2 (en) | 2003-06-03 | 2006-12-12 | Rib-X Pharmaceuticals, Inc. | Biaryl heterocyclic compounds and methods of making and using the same |
| US7705026B2 (en) | 2003-06-03 | 2010-04-27 | Rib-X Pharmaceuticals, Inc. | Biaryl heterocyclic compounds and methods of making and using the same |
| US9550783B2 (en) | 2003-06-03 | 2017-01-24 | Melinta Therapeutics, Inc. | Biaryl heterocyclic compounds and methods of making and using the same |
| US6969726B2 (en) | 2003-06-03 | 2005-11-29 | Rib X Pharmaceuticals Inc | Biaryl heterocyclic compounds and methods of making and using the same |
| US7304050B2 (en) | 2003-09-16 | 2007-12-04 | Pfizer Inc. | Antibacterial agents |
| US7129259B2 (en) | 2003-12-17 | 2006-10-31 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US8202843B2 (en) | 2004-02-27 | 2012-06-19 | Rib-X Pharmaceuticals, Inc. | Macrocyclic compounds and methods of making and using the same |
| US8841263B2 (en) | 2004-02-27 | 2014-09-23 | Melinta Therapeutics, Inc. | Macrocyclic compounds and methods of making and using the same |
| US9376400B2 (en) | 2005-06-08 | 2016-06-28 | Melinta Therapeutics, Inc. | Process for the synthesis of triazoles |
| US8399660B2 (en) | 2005-06-08 | 2013-03-19 | Rib-X Pharmaceuticals, Inc. | Process for the synthesis of triazoles |
| US8796465B2 (en) | 2005-06-08 | 2014-08-05 | Melinta Therapeutics, Inc. | Process for the syntheses of triazoles |
| WO2008038092A3 (en) * | 2006-09-25 | 2009-08-27 | Wockhardt Research Centre | Substituted piperidinophenyl oxazolidinones |
| US8288416B2 (en) | 2006-09-25 | 2012-10-16 | Wockhardt Ltd. | Substituted piperidinophenyl oxazolidinones |
| WO2008038092A2 (en) | 2006-09-25 | 2008-04-03 | Wockhardt Research Centre | Substituted piperidinophenyl oxazolidinones |
| US9572809B2 (en) | 2012-07-18 | 2017-02-21 | Spero Trinem, Inc. | Combination therapy to treat Mycobacterium diseases |
| US9937192B2 (en) | 2012-07-18 | 2018-04-10 | Spero Trinem, Inc. | Combination therapy to treat mycobacterium diseases |
Also Published As
| Publication number | Publication date |
|---|---|
| EE200200282A (en) | 2003-06-16 |
| EP1237895A2 (en) | 2002-09-11 |
| HUP0204052A2 (en) | 2003-03-28 |
| KR20020058072A (en) | 2002-07-12 |
| AR026700A1 (en) | 2003-02-26 |
| CO5271712A1 (en) | 2003-04-30 |
| CZ20021912A3 (en) | 2002-08-14 |
| SK7862002A3 (en) | 2003-02-04 |
| ZA200203876B (en) | 2003-08-15 |
| GB9928499D0 (en) | 2000-02-02 |
| MXPA02005396A (en) | 2002-11-29 |
| IL149641A0 (en) | 2002-11-10 |
| AU762241B2 (en) | 2003-06-19 |
| NO20022605L (en) | 2002-06-26 |
| JP2003515539A (en) | 2003-05-07 |
| IS6401A (en) | 2002-05-28 |
| CA2395052A1 (en) | 2001-06-07 |
| BG106728A (en) | 2003-02-28 |
| CN1433421A (en) | 2003-07-30 |
| WO2001040236A3 (en) | 2002-05-10 |
| BR0016087A (en) | 2002-08-06 |
| PL364762A1 (en) | 2004-12-13 |
| AU1715601A (en) | 2001-06-12 |
| NO20022605D0 (en) | 2002-05-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU762241B2 (en) | Chemical processes and intermediates | |
| JP7030749B2 (en) | Chiral auxiliary | |
| US8470987B2 (en) | Protective group for synthesis of RNA and derivative | |
| US6617339B1 (en) | Oxazolidinone derivatives, process for their preparation and pharmaceutical compositions containing them | |
| FR3037958A1 (en) | ||
| JPH08301869A (en) | Heteroatom-containing benzocyclopentaneoxazolidinone | |
| PL183512B1 (en) | Antibacterial phenyloxazolydinones | |
| KR20050106064A (en) | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents | |
| KR20010085887A (en) | Heterocyclyl amino methyloxa zolidinones as antibacterials | |
| US20210317159A1 (en) | Alkoxyphenyl derivatives, protected nucleosides and protected nucleotides, method for producing oligonucleotides, and method for removing substituents | |
| EP3560925A1 (en) | Novel compound and pharmaceutically-acceptable salt thereof | |
| JP2008500318A (en) | 3- {4- (Pyridin-3-yl) phenyl} -5- (1H-1,2,3-triazol-1-ylmethyl) -1,3-oxazolidine-2-one as an antibacterial agent | |
| MXPA06013540A (en) | 3- (4- (2-dihydroisoxazol-3-ylpyridin-5-yl) phenyl) -5-triazol-1-ylmethyloxazolidin-2-one derivaives as mao inhibitors for the treatment of bacterial infections. | |
| WO2002096916A1 (en) | Process for phosphorylation | |
| JPH09500131A (en) | Method for producing HIV protease inhibitor | |
| JPH10506621A (en) | Intermediates for dinucleotide and oligonucleotide analogs | |
| WO2004048370A1 (en) | Antibacterial compounds | |
| JP2006520774A (en) | Antibacterial 1,3-oxazolidine-2-one derivative | |
| JP2006520775A (en) | Antibacterial oxalidinones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 149641 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/03876 Country of ref document: ZA Ref document number: 200203876 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 17156/01 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000979764 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2000 106728 Country of ref document: BG Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IN/PCT/2002/00641/MU Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 519083 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2395052 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/005396 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2002-1912 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027007072 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2001 540991 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 7862002 Country of ref document: SK |
|
| ENP | Entry into the national phase |
Ref document number: 2002 2002117647 Country of ref document: RU Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027007072 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 008187355 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2002-1912 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000979764 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000979764 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 17156/01 Country of ref document: AU |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV2002-1912 Country of ref document: CZ |