WO1999043672A1 - Inhibitors of phospholipase a2 - Google Patents
Inhibitors of phospholipase a2 Download PDFInfo
- Publication number
- WO1999043672A1 WO1999043672A1 PCT/US1999/003388 US9903388W WO9943672A1 WO 1999043672 A1 WO1999043672 A1 WO 1999043672A1 US 9903388 W US9903388 W US 9903388W WO 9943672 A1 WO9943672 A1 WO 9943672A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- cooh
- aikoxy
- compound
- benzyl
- Prior art date
Links
- 0 *C1c(cccc2)c2N(*)*1 Chemical compound *C1c(cccc2)c2N(*)*1 0.000 description 11
- JUJWROOIHBZHMG-UHFFFAOYSA-N c1ccncc1 Chemical compound c1ccncc1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- YXFVVABEGXRONW-UHFFFAOYSA-N Cc1ccccc1 Chemical compound Cc1ccccc1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- IMRZMNPKDMFTBV-NSCUHMNNSA-N C/C=C(\C(N1)=O)/SC1=O Chemical compound C/C=C(\C(N1)=O)/SC1=O IMRZMNPKDMFTBV-NSCUHMNNSA-N 0.000 description 1
- XSXFYGXACNARRT-XNWCZRBMSA-N C/C=C(\C(N1c2ccccc2)=O)/SC1=O Chemical compound C/C=C(\C(N1c2ccccc2)=O)/SC1=O XSXFYGXACNARRT-XNWCZRBMSA-N 0.000 description 1
- CDRZEVAFRDEIEJ-UHFFFAOYSA-N CC(N[S+2](C)=O)O Chemical compound CC(N[S+2](C)=O)O CDRZEVAFRDEIEJ-UHFFFAOYSA-N 0.000 description 1
- XQIAIYRFTRMNKM-UHFFFAOYSA-O CC(N[S+](C)(C)=O)=O Chemical compound CC(N[S+](C)(C)=O)=O XQIAIYRFTRMNKM-UHFFFAOYSA-O 0.000 description 1
- YMKFJVYMLXCNCH-UHFFFAOYSA-P CC(N[S+](O)(I)=O)=O Chemical compound CC(N[S+](O)(I)=O)=O YMKFJVYMLXCNCH-UHFFFAOYSA-P 0.000 description 1
- WZBHZJBLEBPCSB-UHFFFAOYSA-O CC(N[SH+](C)=O)=O Chemical compound CC(N[SH+](C)=O)=O WZBHZJBLEBPCSB-UHFFFAOYSA-O 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N Cc1ncccc1 Chemical compound Cc1ncccc1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- ARJRVMQOHCHFEA-UHFFFAOYSA-N Cc1sc(cccc2)c2[nH]1 Chemical compound Cc1sc(cccc2)c2[nH]1 ARJRVMQOHCHFEA-UHFFFAOYSA-N 0.000 description 1
- UHOVQNZJYSORNB-UHFFFAOYSA-N c1ccccc1 Chemical compound c1ccccc1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/32—Oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/12—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/18—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D209/22—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with an aralkyl radical attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/42—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/86—Oxygen and sulfur atoms, e.g. thiohydantoin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/34—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/79—Benzo [b] furans; Hydrogenated benzo [b] furans with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
- C07D307/81—Radicals substituted by nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/14—Radicals substituted by singly bound hetero atoms other than halogen
- C07D333/20—Radicals substituted by singly bound hetero atoms other than halogen by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
Definitions
- the present invention relates to chemical inhibitors of the activity of various phospholipase enzymes, particularly phospholipase A enzymes.
- Leukotrienes and prostaglandins are important mediators of inflammation. Leukotrienes recruit inflammatory cells such as neutrophils to an inflamed site, promote the extravasation of these cells and stimulate release of superoxide and proteases which damage the tissue. Leukotrienes also play a pathophysiological role in the hypersensitivity experienced by asthmatics [See, e.g. B. Samuelson et al., Science. 237: 1171-76 (1987)]. Prostaglandins enhance inflammation by increasing blood flow and therefore infiltration of leukocytes to inflamed sites. Prostaglandins also potentiate the pain response induced by stimuli.
- Prostaglandins and leukotrienes are unstable and are not stored in cells, but are instead synthesized [W. L. Smith. Biochem. J.. 259:315-324 (1989)] from arachidonic acid in response to stimuli.
- Prostaglandins are produced from arachidonic acid by the action of COX-1 and COX-2 enzymes.
- Arachidonic acid is also the substrate for the distinct enzyme pathway leading to the produciton of leukotrienes.
- PLA 2 phospholipase A
- the reaction catalyzed by PLA is believed to represent the rate-limiting step in the process of lipid mediated biosynthesis and the production of inflammatory prostaglandins and leukotrienes.
- PAF platelet activating factor
- anti-inflammatory therapies have focussed on preventing production of either prostaglandins or leukotrienes from these distinct pathways, but not on all of them.
- ibuprofen, aspirin and indomethacin are all NSAIDs which inhibit the production of prostaglandins by COX-l/COX-2, but have no effect on the inflammatory production of leukotrienes from arachidonic acid in the other pathways.
- zileuton inhibits only the pathwasy of conversion of arachidonic acid to leukotrienes, witout affecting the production of prostaglandins. None of these widelt-used anti- inflammatory agents affects the production of PAF.
- This non- pancreatic PLA is found in platelets, synovial fluid, and spleen and is also a secreted enzyme. This enzyme is a member of the aforementioned family. [See, J. J. Seilhamer et al. J. Biol. Chem.. 264:5335-5338 (1989); R. M. Kramer et al. J. Biol. Chem.. 264:5768-5775 (1989); and A. Kando et al, Biochem. Biophys. Res. Comm.. 163:42-48 (1989)].
- a murine PLA 2 has been identified in the murine macrophage cell line, designated RAW 264.7. A specific activity of 2 ⁇ mols/min/mg, resistant to reducing conditions, was reported to be associated with the approximately 60 kD molecule. However, this protein was not purified to homogeneity. [See, C. C. Leslie et al, Biochem. Biophvs. Acta.. 963:476-492 (1988)]. The references cited above are incorporated by reference herein for information pertaining to the function of the phospholipase enzymes, particularly PLA,.
- cytosolic phospholipase A (hereinafter "cPLA,") has also been identified and cloned. See, U.S. Patent Nos. 5,322,776 and 5,354,677, which are incorporated herein by reference as if fully set forth.
- the enzyme of these patents is an intracellular PLA enzyme, purified from its natural source or otherwise produced in purified form, which functions intracellularly to produce arachidonic acid in response to inflammatory stimuli.
- the present invention provides compounds having a chemical formula selected from the group consisting of:
- A is independent of any other group and is selected from the group consisting of -CH 2 - and -CH 2 -CH 2 -;
- R is independent of any other R group and is selected from the group consisting of -X-R, -H. - OH, halogen, -CN, -NO,, C,-C 5 alkyl, alkenyl, alkinyl, aryl and substituted aryl;
- R 2 is independent of any other R group and is selected from the group consisting of -H, -COOH, -COR 5 , -CONR ⁇ , -(CH 2 ) n -W-(CH 2 ) m -Z-R 5 , -(CH 2 ) n -W-R 5 , -Z-R 5 , C,-C 10 alkyl, alkenyl and substimted aryl;
- R 3 is independent of any other R group and is selected from the group consisting of -H, -COOH, -COR 5 , -CONR 5 R «, -(CH 2 ) n -W-(CH 2 ) m -Z-R 5 , -(CH 2 ) n -W-R 5 , -Z-R 5 , C,-C 10 alkyl, alkenyl and substituted aryl;
- R 4 is independent of any other R group and is selected from the group consisting of -H, -OH, - OR,, -SRj, -CN, -COR ⁇ , -NHR 6 , -COOH, -CONR ⁇ , -NO,, -CONHS0 2 R 8 , C,-C 5 alkyl, alkenyl and substituted aryl;
- R 5 is independent of any other R group and is selected from the group consisting of -H, -OH, 0(CH,) n R 6 , -SR ⁇ , -CN, -COR,, -NHR,, -COOH, -NO,, -COOH, -CONR,R 7 , -CONHSO,R 8 , C,-C 5 alkyl, alkenyl, alkinyl, aryl, substituted aryl, -CF 3 , -CF,CF 3 and
- R is independent of any other R group and is selected from the group consisting of -H, C-C 5 alkyl, alkenyl, alkinyl, aryl and substituted aryl;
- R 7 is independent of any other R group and is selected from the group consisting of -H, C-C 5 alkyl, alkenyl, alkinyl, aryl and substituted aryl;
- R 8 is independent of any other R group and is selected from the group consisting of C-C 3 alkyl, aryl and substituted aryl;
- Rt is independent of any other R group and is selected from the group consisting of -H, -OH, a halogen, -CN, -OR,, -COOH, -CONR,R 7 , tetrazole, -CONHSO,R 8 , -COR,, -(CH,) interceptCH(OH)R 6 and -(CH,) n CHR 6 R 5 ;
- Rio is independent of any other R group and is selected from the group consisting of -H, -OH, a halogen, -CN, -OR,, -COOH, -CONR,R 7 , tetrazole, -CONHSO,R 8> -COR,, -(CH,) n CH(OH)R, and -(CH,) n CHR,R 5 ;
- X is independent of any other group and is, independently each time used including within the same compound, selected from the group consisting of -0-, -S- and -N(R6)-;
- Z is independent of any other group and is, independently each time used including within the same compound, selected from the group consisting of -CH-, -0-, -S-, -N(R,)-, -CO-, -CON(R,)- and - N(R,)CO-;
- m is, independently each time used including within the same compound, an integer from 0 to 4; and
- n is independent of m and is, independently each time used including within the same compound, an integer from 0 to 4.
- the compounds of the invention have phospholipase enzyme inhibiting activity.
- Other preferrred embodiments include compounds having the following chemical formula:
- A is -CH- and R, is -(CH 2 ) n -W-(CH,) m -ZR 5
- R is -(CH 2 ) n -W-(CH,) m -ZR 5
- n is 1
- m is 1
- W is -S- and Z is -CO-
- Rj is -NHR
- R is a substituted aryl group and those wherein said aryl group is substituted with one or more substituents independently selected from the group consisting of a halogen, -CF 3 ,
- R is selected froup the group consisting of alkyl, alkenyl, alkynyl, -(CH,) p OH, and -0(CH 2 ) p CH 3 , and wherein p is an integer from 0 to 4.
- R is selected from the group consisting of -H and -OCH(C 6 H 6 ) and R 3 is -COR 5 , R 5 is -OC ⁇ R, and R, is a substituted aryl group.
- said aryl group is substimted with one or more substituents selected from the group consisting of -CF, - CF,CF 3 and -C(CH 3 ),CH,CH 3 .
- R, and R r are independently selected from C,-C 6 alkyl, -Z-C j -C 5 alkyl, phenyl, -(CH 2 ) n - Z-(CH 2 ) n -phenyl, benzyl, -(CH 2 ) n -Z-(CH 2 ) n -benzyl, napthyl, -(CH 2 ) n -Z-(CH 2 ) n -napthyl, pyrimidinyl, -(CH 2 ) n -Z-(CH 2 ) n -pyrimidinyl, the alkyl, phenyl, benzyl, napthyl and pyrimidinyl groups being optionally substituted by from 1 to 3 substituents selected from halogen, C,-C 6 alkyl, C,-C 6 aikoxy, -NO 2 , -NH,, -CN, -CF 3 , or -
- Z is O or S
- n is an integer from 0 to 3;
- R 2 is selected from H, halogen, -CF 3 , -OH, -C,-C 10 alkyl, C,-C 10 aikoxy, -CHO, -CN, - NO 2 , -NH 2 , -NH-C,-C 6 alkyl, -N(C,-C 6 alkyl) 2 , -N-SO 2 -C r C 6 alkyl, or -SO 2 -C,-C 6 alkyl;
- R 3 is selected from H, halogen, -CF 3 , -OH, -C,-C 10 alkyl, C,-C 10 aikoxy, -CHO, - C(O)CH 3 , -C(O)-(CH 2 )n-CF 3 , -CN, -NO 2 , -NH 2 , -NH-C r C 6 alkyl, -N(C,-C 6 alkyl),, -N-SO 2 - C,-C 6 alkyl, -SO,-C,-C 6 alkyl or a moiety of the formula:
- n in each appearance is independently selected as an integer selected from 0-3;
- R 8 and R 9 are independently selected in each appearance from H, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) ⁇ -C(O)-COOH, -CF 3 , -OH, -(CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,-C 6 alkyl, -NH(C r C 6 alkyl), or -N(C,-C 6 alkyl) 2 ;
- R is selected from H, -CF 3 , C,-C 6 alkyl, -(CH 2 ) n -C 3 -C 6 cycloalkyl, phenyl, or benzyl, the cycloalkyl, phenyl or benzyl groups being optionally substituted by from 1 to 3 groups selected from halogen, -CF 3 , -OH, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,- C 6 alkyl, -NH(C,-C 6 alkyl), or -N(C,-C 5 alkyl) 2 ;
- L 1 is selected from -(CH 2 ) n -O-, -(CH 2 ) n -S-, -(CH 2 ) compassion-O-(CH 2 ) n -, -(CH 2 ) n -S-(CH 2 ) n -, -C(O)-O-, -C(O)-(CH 2 ) n -O-, -C(O)-N-, or -(CH 2 ) n -S-(CH 2 ) n -C(O)-N-;
- M' is -COOH or a moiety selected from:
- R 10 is selected from H, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -CF 3 , -OH, (CH 2 ) ⁇ -C(O)-COOH, -C,-C 6 alkyl, -O-C,-C 6 alkyl,
- R 5 is selected from:
- L 2 is selected from a chemical bond or a bridging group selected from -(CH 2 ) ⁇ -Z-, -(CH 2 ) n -Z-(CH 2 ) n -, -C(O)-O-, -C(O)-(CH 2 ) n -O-, -C(O)-N-, or -(CH 2 ) ⁇ -S-(CH 2 ) n -C(O)-N-;
- M 2 is selected from -C,-C 6 alkyl, -O-C,-C 6 alkyl,
- R 8 and R 9 are as defined above and can be substituted anywhere on the cyclic or bicyclic ring; or
- L 3 is a chemical bond or a group selected from -CH 2 - , -CH 2 -Z- , -C(O)- , -O-, -S- , or -(CH 2 ) n -Z-(CH 2 ) n -;
- M 3 is selected from -(CH 2 ) n -C 3 -C 5 cycloalkyl, furanyl, thienyl, pyrrolyl,
- a preferred subset include those in which the core molecule is an indole.
- R' and R 2 are hydrogen, and the moieties R 3 , R 4 , R 5 , R 8 , R 9 and R 10 , n, L 1 , L 2 , M 1 and M 2 are as defined above.
- R 1 is in the indole 5-position.
- R is selected from -O-C,-C 6 alkyl, -S-C r C 6 alkyl, -O-phenyl, -S-phenyl, -O-benzyl, -S- benzyl, the alkyl, phenyl or benzyl groups being optionally substituted by from 1 to 3 substituents selected from halogen, C,-C 6 alkyl, C,-C 6 aikoxy, -NO 2 , -NH 2 , -CN, -CF literal or -OH;
- R 2 is selected from H, halogen, -CF 3 , -OH, -C,-C 10 alkyl, preferably -C,-C 6 alkyl, C,-C I0 aikoxy, preferably C,-C 6 aikoxy, -CHO, -CN, -NO 2 , -NH 2 , -NH-C,-C 6 alkyl, -N(C,-
- R 3 is selected from H, halogen, -CF 3 , -OH, -C,-C 10 alkyl, preferably -C r C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -CN, -NO 2 , -NH 2 , -NH-C r C 6 alkyl, -N(C,-C 6 alkyl) 2 , - N-80 2 -C j -Cg alkyl, -SO j -C ⁇ alkyl, or a moiety of the formula:
- n in each appearance is independently selected as an integer selected from 0-3;
- R 8 and R 9 are independently selected in each appearance from H, -COOH, -(CH 2 ) n -COOH, -(CH,) n -C(O)-COOH, -CF 3 , -OH, -(CH,) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,-C 6 alkyl, -NH(C,- C 6 alkyl), or -N(C,-C 6 alkyl) 2 ;
- R 4 is the moiety -L'-M 1 or
- L 1 is selected from a chemical bond or a bridging group selected from -(CH 2 ) n -O-, -(CH,) n -S-, -(CH,) n -O-(CH,) n -, -(CH 2 ) n -S-(CH 2 ) n -, -C(O)-O-, -C(O)-(CH,_) n -O-, -C(O)-N-, or -(CH 2 ) n -S-(CH,) n -C(O)-N-;
- M is the moiety:
- R ⁇ is selected from H, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -CF 3 , -OH, (CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,-C 6 alkyl,
- R is a structure of the formula
- L 2 is selected from a chemical bond or a bridging group selected from -(CH 2 ) n -O-, -(CH 2 ) ⁇ -S-, -(CH 2 ) ⁇ -O-(CH 2 ) n -, -(CH 2 ) n -S-(CH 2 ) n -, -C(O)-O-, -C(O)-(CH 2 ) n -O-, -C(O)-N-, or -(CH 2 ) n -S-(CH,) n -C(O)-N-;
- M 2 is selected from -C,-C 6 alkyl, -O-C,-C 6 alkyl,
- R 8 , R 9 and R 10 are as defined above; or a pharmaceutically acceptable salt thereof.
- R is selected from -O-C,-C 6 alkyl, -S-C,-C 6 alkyl, -O-phenyl, -O-benzyl, -S-benzyl, the alkyl, phenyl or benzyl groups being optionally substituted by from 1 to 3 substituents selected from halogen, C,-C 6 alkyl, C,-C 6 aikoxy, -NO 2 , -NH 2 , -CN, -CF 3 , or -OH;
- R 3 is selected from H, halogen, -CF 3 , -OH, -C,-C 10 alkyl, preferably -C ⁇ -C 10 alkyl, C,-C, 0 aikoxy, preferably C,-C I0 aikoxy, -CHO, -CN, -NO 2 , -NH 2 , -NH-C,-C 6 alkyl, -N(C,-C 6 alkyl) 2 , -N-SO 2 -C,-C 6 alkyl, -SO 2 -C,-C 6 alkyl or a moiety of the formula:
- R , R , R , R and R are as defined above, or a pharmaceutically acceptable salt thereof.
- R, and R r are independently selected from H, halogen, -CF 3 , -OH, -C,-C 10 alkyl, preferably -C,-C 6 alkyl, -S-C,-C I0 alkyl, preferably -S-C,-C 6 alkyl, C,-C I0 aikoxy, preferably C,- C 6 aikoxy, -CN, -NO 2 , -NH 2 , phenyl, -O-phenyl, -S-phenyl, benzyl, -O-benzyl, -S-benzyl; or a ring moiety of the groups a), b) or c), below, directly bonded to the indole ring or bonded to the indole ring by a -S-, -O- or -(CH 2 ) n - bridge;
- a bicyclic ring moiety optionally containing from 1 to 3 ring heteroatoms selected from N, S or O including, but not limited to benzofuran, chromene, indole, isoindole, indoline, isoindoline, napthalene, purine, indolizine, indazole, quinoline, isoquinoline, quinolizine, quinazoline, cinnoline, phthalazine, or napthyridine, the bicyclic ring moiety being optionally substituted by from 1 to 3 substituents selected from halogen, C r C 10 alkyl, preferably C r C 6 alkyl, C r C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -NO 2 , -NH 2 , -CN, -CF 3 or -OH; or
- Z is O or S
- R 6 is selected from the relevant members of the group H, -CF 3 , C,-C 10 alkyl, preferably C,-C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, phenyl, -O-phenyl, -S-phenyl, benzyl, -O- benzyl, or -S-benzyl, the phenyl and benzyl rings of these groups being optionally substituted by from 1 to 3 substituents selected from halogen, C,-C 10 alkyl, preferably C C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -NO 2 , -NH 2 , -CN, -CF 3 , or -OH;
- R 7 is selected from the relevant members of the group -OH, -CF 3 , C ⁇ C ⁇ alkyl, preferably C r C 6 alkyl, C,-C 10 aikoxy, preferably C r C 6 aikoxy, -NH 2 , -(CH 2 ) n -NH 2 , -NH-(C,-C 6 alkyl), - N-(C,-C 6 alkyl) 2 , -(CH 2 ) n -NH-(C,-C 6 alkyl), -(CH 2 ) conflict-N-(C,-C 6 alkyl) 2 , phenyl, -O-phenyl, benzyl, or -O-benzyl; or
- a bicyclic ring moiety containing from 8 to 10 ring atoms and optionally containing from 1 to 3 ring heteroatoms selected from N, S or O including, but not limited to benzofuran, chromene, indole, isoindole, indoline, isoindoline, napthalene, purine, indolizine, indazole, quinoline, isoquinoline, quinolizine, quinazoline, cinnoline, phthalazine, or napthyridine, the bicyclic ring moiety being optionally substituted by from 1 to 3 substituents selected from halogen, C,-C 10 alkyl, preferably C r C 6 alkyl, C r C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -NO 2 , - NH 2 , -CN, -CF 3 or -OH;
- n is an integer from 0 to 3, preferably 1 to 3, more preferably 1 to 2;
- R 2 is selected from H, halogen, -CN, -CHO, -CF 3 , -OH, C,-C 10 alkyl, preferably C,-C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -CN, -NO 2 , -NH 2 , -NH-C.-C, alkyl, -N(C,-C 6 alkyl),, -N-SO,-C,-C 6 alkyl, or -SO 2 -C,-C 6 alkyl;
- R 3 is selected from H, halogen, -CF 3 , -OH, -C,-C I0 alkyl, C,-C 10 aikoxy, -CHO, - C(O)CH 3 , -C(O)-(CH 2 )n-CF 3 , -CN, -NO 2 , -NH 2 , -NH-C.-C
- n in each appearance is an integer independently selected from 0-3;
- R 8 and R 9 are independently selected in each appearance from H, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -CF 3 , -OH, -(CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C r C 6 alkyl, -NH(C,- C 6 alkyl), or -N(C r C 6 alkyl) 2 ;
- R 12 is selected from H, -CF 3 , C,-C 6 alkyl, -(CH 2 ) n -C 3 -C 6 cycloalkyl, phenyl, or benzyl, the cycloalkyl, phenyl or benzyl groups being optionally substituted by from 1 to 3 groups selected from halogen, -CF 3 , -OH, -COOH, -(CH 2 ) ⁇ -COOH, -(CH 2 ) ⁇ -C(O)-COOH, -C,-C 6 alkyl, -O-C,- C 6 alkyl, -NH(C,-C 6 alkyl), or -N(C,-C 6 alkyl) 2 ;
- L 1 is selected from -(CH 2 ) n -, -S-, -O-, -C(O)-, -C(O)-O-,-(CH 2 ) n -O-, -(CH 2 ) n -S-, -(CH 2 ) n -O-(CH 2 ) n -, -(CH 2 ) n -S-(CH 2 ) n -, -(CH 2 ) n -C(O)-(CH 2 ) n -, -(CH 2 ) n -O-(CH 2 ) n -, -(CH 2 ) ⁇ -S-(CH 2 ) n -,-C(Z)-N(R 6 )-(CH 2 ) n -, -C(O)-C(Z)-N(R 6 )-, -C(O)-N(R 6 )-, -C(O)
- M 1 is -COOH or a moiety selected from:
- R 8 in each appearance, is independently selected from H, -COOH, -(CH 2 ) n -COOH, (CH 2 ) n -C(O)-COOH, tetrazole,
- R 9 in each appearance is independently selected from H, halogen, -CF 3 , -OH, -COOH, (CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,-C 6 alkyl, -NH(C r C 6 alkyl), or -N(C,-C 6 alkyl) 2 ;
- R 10 is selected from H, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -CF 3 , -OH, (CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C.-C, alkyl,
- R Thallium is selected from H, C,-C 6 lower alkyl, C,-C 6 cycloalkyl, -CF 3 , -COOH, -(CH 2 ) n - COOH, -(CH 2 ) n -C(O)-COOH,
- the moiety or combination of moieties comprising R 4 include an acidic group selected from carboxylic acid, a tetrazole or a moiety of the formulae:
- R 5 is selected from C r C 6 lower alkyl, C,-C 6 lower aikoxy, -(CH 2 ) n -C 3 -C 10 cycloalkyl, -(CH 2 ) n -S-(CH 2 ) n -C 3 -C 10 cycloalkyl, -(CH 2 ) n -O-(CH 2 ) n -C 3 -C 10 cycloalkyl, or the groups of:
- n is an integer from 0 to 3, preferably 1 to 3, more preferably 1 to 2,
- Y is C 3 -C 5 cycloalkyl
- a bicyclic ring moiety containing from 8 to 10 ring atoms and optionally containing from 1 to 3 ring heteroatoms selected from N, S or O including, but not limited to benzofuran, chromene, indole, isoindole, indoline, isoindoline, napthalene, purine, indolizine, indazole, quinoline, isoquinoline, quinolizine, quinazoline, cinnoline, phthalazine, or napthyridine, the bicyclic ring moiety being optionally substituted by from 1 to 3 substituents selected from halogen, C,-C 10 alkyl, preferably C r C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -NO,, - NH 2 , -CN, -CF 3 or -OH;
- D is H, C,-C 6 lower alkyl, C,-C 5 lower aikoxy, -CF 3 or -(CH 2 ) n -CF 3 ;
- B and C are independently selected from phenyl, pyridinyl, pyrimidinyl, furyl, thienyl or pyrrolyl groups, each optionally substituted by from 1 to 3, preferably 1 to 2, substituents selected from H, halogen, -CN, -CHO, -CF 3 , -OH, -C,-C 6 alkyl, C,-C 6 aikoxy, -NH, or -NO 2 ; or a pharmaceutically acceptable salt thereof.
- Preferred compounds include those having the formula:
- R j is selected from H, halogen, -CF 3 , -OH, -C,-C 10 alkyl, preferably -C,-C 6 alkyl, -S-C,- C 10 alkyl, preferably -S-C,-C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -CN, -NO,, -NH 2 , phenyl, -O-phenyl, -S-phenyl, benzyl, -O-benzyl, -S-benzyl; or a ring moiety of the groups a), b) or c), below, directly bonded to the indole ring or bonded to the indole ring by a -S-, -O- or - (CH 2 ) n - bridge;
- furan, pyrrole, or thiophene being optionally substituted by from 1 to 3 substituents selected from halogen, C r C 10 alkyl, preferably C,-C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -NO 2 , -NH 2 , -CN, -CF 3 ; or
- pyridine, pyrimidine, piperidine, or morpholine each being optionally substituted by from 1 to 3 substituents selected from halogen, C,-C 10 alkyl, preferably C,-C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -NO 2 , -NH,, -CN, -CF, or -OH; or
- Z is O or S
- R 6 is selected from the relevant members of the group H, -CF 3 , C,-C 10 alkyl, preferably C,-C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, phenyl, -O-phenyl, -S-phenyl, benzyl, -O- benzyl, or -S-benzyl, the phenyl and benzyl rings of these groups being optionally substituted by from 1 to 3 substituents selected from halogen, C r C 10 alkyl, preferably C,-C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -NO,, -NH 2 , -CN, -CF 3 , or -OH;
- R 7 is selected from the relevant members of the group -OH, -CF 3 , C,-C 10 alkyl, preferably C r C 6 alkyl, C,-C I0 aikoxy, preferably C,-C 6 aikoxy, -NH 2 , -(CH 2 ) n -NH 2 , -NH-(C r C 6 alkyl), - N-(C,-C 6 alkyl) 2 , -(CH 2 ) n -NH-(C r C 6 alkyl), -(CH 2 ) n -N-(C,-C 6 alkyl) 2 , phenyl, -O-phenyl, benzyl, or -O-benzyl, furan, pyrrole, thiophene, pyridine, pyrimidine, thiazole, pyrazole, or morpholine the rings of these groups being optionally substituted by from 1 to 3 substituents selected from hal
- R 2 is selected from H, halogen, -CN, -CHO, -CF 3 , -OH, C,-C 10 alkyl, preferably C,-C 6 alkyl, C,-C 10 aikoxy, preferably C r C 6 aikoxy, -CHO, -CN, -NO 2 , -NH 2 , -NH-C,-C 6 alkyl, -N(C,-C 6 alkyl) 2 , -N-SO 2 -C,-C 6 alkyl, or -SO 2 -C,-C 6 alkyl;
- R 3 is selected from H, halogen, -CF 3 , -OH, -C r C 10 alkyl, C,-C 10 aikoxy, -CHO, -C(O)CH 3 , -C(O)-(CH 2 )n-CF 3 , -CN, -NO 2 , -NH 2 , -NH-C r C 6 alkyl, -N(C,-C 6 alkyl) 2 , -N-SO 2 - C,-C 6 alkyl, -SO 2 -C,-C 6 alkyl, phenyl, phenyloxy, benzyl, benzyloxy-C(O)-phenyl, -C(O)- benzyl, -CH 2 -(C 3 -C 5 cycloalky), -C(O)-OH, C(O)-C r C 6 alkyl, -C(O)-O-C r C 6 alkyl
- n in each appearance is independently selected as an integer selected from 0-3;
- R 8 and R 9 are independently selected in each appearance from H, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -CF 3 , -OH, -(CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C r C 6 alkyl, -NH(C r C 6 alkyl), or -N(C,-C 6 alkyl),_;
- R 12 is selected from H, -CF 3 , C,-C 6 alkyl, -(CH 2 ) n -C 3 -C 6 cycloalkyl, phenyl, or benzyl, the cycloalkyl, phenyl or benzyl groups being optionally substituted by from 1 to 3 groups selected from halogen, -CF 3 , -OH, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,- C 6 alkyl, -NH(C,-C 6 alkyl), or -N(C,-C 6 alkyl) 2 ;
- L 1 is selected from -(CH 2 ) n -, -S-, -O-, -C(O)-, -C(O)-O-,-(CH 2 ) n -O-, -(CH 2 ) n -S-, -(CH 2 ) n -O-(CH 2 ) n -, -(CH 2 ) n -S-(CH 2 ) n -, -(CH 2 ) ⁇ -C(O)-(CH 2 ) n -, -(CH,) n -O-(CH 2 ) n -, -(CH 2 ) n -S-(CH 2 ) n -,-C(Z)-N(R 6 K -C(Z)-N(R 6 )-(CH 2 ) n -, -C(O)-C(Z)-N(R 6 )-, -C(O)-C(Z
- M' is -COOH or a moiety selected from:
- R 8 in each appearance, is independently selected from H, -COOH, -(CH 2 ) n -COOH, (CH,) n -C(O)-COOH, tetrazole,
- R 9 in each appearance is independently selected from H, halogen, -CF 3 , -OH, -COOH, - (CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,-C 6 alkyl, -NH(C,-C 6 alkyl), or -N(C,-C 6 alkyl) 2 ;
- R 10 is selected from H, -COOH, -(CH,) n -COOH, -(CH,) n -C(O)-COOH, -CF 3 , -OH, - (CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C r C 6 alkyl,
- the moiety or combination of moieties comprising R 4 include an acidic group selected from carboxylic acid, a tetrazole or a moiety of the formulae:
- R 5 is selected from C r C 6 lower alkyl, C r C 6 lower aikoxy, -(CH 2 ) n -C 3 -C 10 cycloalkyl, -(CH 2 ) n -S-(CH 2 ) n -C 3 -C 10 cycloalkyl, -(CH 2 ) n -O-(CH 2 ) n -C 3 -C 10 cycloalkyl, -(CH 2 ) n -phenyl-O- phenyl, -(CH 2 ) n -phenyl-CH 2 -phenyl, -(CH 2 ) n -O-phenyl-CH 2 - ⁇ henyl, -(CH 2 ) n -phenyl-(O-CH 2 - phenyl) 2 , -CH 2 -phenyl-C(O)-benzothiazole or a moiety of the formulae -(
- D is H, C,-C 6 lower alkyl, C,-C 6 lower aikoxy, -CF 3 or -(CH 2 ) n -CF 3 ;
- B and C are independently selected from phenyl, pyridinyl, pyrimidinyl, furyl, thienyl or pyrrolyl groups, each optionally substituted by from 1 to 3, preferably 1 to 2, substituents selected from H, halogen, -CN, -CHO, -CF 3 , -OH, -C r C 6 alkyl, C r C 6 aikoxy, -NH 2 or -NO 2 ; or a pharmaceutically acceptable salt thereof.
- R is selected from H, halogen, -CF 3 , -OH, -C,-C I0 alkyl, preferably -C,-C 6 alkyl, -S-C,- C 10 alkyl, preferably -S-C,-C 5 alkyl, C,-C I0 aikoxy, preferably C,-C 6 aikoxy, -CN, -NO 2 , -NH 2 , phenyl, -O-phenyl, -S-phenyl, benzyl, -O-benzyl, -S-benzyl; or furan, pyrrole, or thiophene, bonded to the indole ring by a chemical bond or a -S-, -O- or -(CH 2 ) n - bridge, the phenyl, benzyl, furan, pyrrole, or thiophene rings being optionally substituted by from 1 to 3 substituents selected from
- n is an integer from 0 to 3, preferably 1 to 3, more preferably 1 to 2;
- R 2 is selected from H, halogen, -CN, -CHO, -CF 3 , -OH, C,-C 10 alkyl, preferably C,-C 6 alkyl, C,-C 10 aikoxy, preferably C,-C 6 aikoxy, -CHO, -CN, -NO 2 , -NH 2 , -NH-C,-C 6 alkyl, -N(C r C 6 alkyl) 2 , -N-SO 2 -C,-C 6 alkyl, or -SO 2 -C,-C 6 alkyl;
- R 3 is selected from H, halogen, -CF 3 , -OH, -C,-C 10 alkyl, C r C 10 aikoxy, -CHO, -C(O)CH 3 , -C(O)-(CH 2 )n-CF 3 , -CN, -NO 2 , -NH 2 , -NH-C,-C 6 alkyl, -N(C,-C 6 alkyl),, -N-SO,- C,-C 6 alkyl, -SO 2 -C,-C 6 alkyl, phenyl, phenyloxy, benzyl, benzyloxy-C(O)-phenyl, -C(O)- benzyl, -CH 2 -(C 3 -C 5 cycloalky), -C(O)-OH, C(O)-C,-C 6 alkyl, -C(O)-O-C,-C 6 alkyl, -
- n in each appearance is independently selected as an integer selected from 0-3;
- R 8 and R 9 are independently selected in each appearance from H, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -CF 3 , -OH, -(CH 2 ) deliberately-C(O)-COOH, -C,-C 6 alkyl, -O-C r C 6 alkyl, -NH(C,- C 6 alkyl), or -N(C,-C 6 alkyl) 2 ;
- R is selected from H, -CF 3 , C,-C 6 alkyl, -(CH 2 ) n -C 3 -C 6 cycloalkyl, phenyl, or benzyl, the cycloalkyl, phenyl or benzyl groups being optionally substituted by from 1 to 3 groups selected from halogen. -CF 3 , -OH. -COOH, -(CH,) n -COOH, -(CH,) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,- C 6 alkyl, -NH(C,-C 6 alkyl), or -N(C,-C 6 alkyl) 2 ;
- L 1 is selected from -(CH,) n -, -S-, -O-, -C(O)-, -C(O)-O-,-(CH 2 ) n -O-, -(CH 2 ) n -S-, -(CH 2 ) n -O-(CH 2 ) n -, -(CH 2 ) n -S-(CH 2 ) n -, -(CH,) n -C(O)-(CH 2 ) n -, -(CH 2 ) n -O-(CH 2 ) n -, -(CH 2 ) ⁇ -S-(CH 2 ) n -,-C(Z)-N(R 6 )-, -C(O)-C(Z)-N(R 6 )-, -C(O)-C(Z)-N(R 6 )-, -C(O)-C(Z)-
- M 1 is -COOH or a moiety selected from:
- R 8 in each appearance, is independently selected from H, -COOH, -(CH 2 ) ⁇ -COOH, - (CH 2 ) n -C(O)-COOH, tetrazole,
- R 9 in each appearance is independently selected from H, halogen, -CF 3 , -OH, -COOH, - (CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,-C 6 alkyl, -NH(C,-C 6 alkyl), or -N(C,-C 6 alkyl),;
- R 10 is selected from H, -COOH, -(CH 2 ) n -COOH, -(CH 2 ) n -C(O)-COOH, -CF 3 , -OH, - (CH 2 ) n -C(O)-COOH, -C,-C 6 alkyl, -O-C,-C 6 alkyl,
- the moiety or combination of moieties comprising R 4 include an acidic group selected from carboxylic acid, a tetrazole or a moiety of the formulae:
- R 5 is selected from C,-C 6 lower alkyl, C,-C 6 lower aikoxy, -(CH 2 ) n -C 3 -C 10 cycloalkyl, -(CH 2 ) n -S-(CH 2 ) n -C 3 -C 10 cycloalkyl, -(CH 2 ) n -O-(CH 2 ) n -C 3 -C 10 cycloalkyl, -(CH 2 ) n -phenyl-O- phenyl, -(CH 2 ) n -phenyl-CH 2 -phenyl, -(CH 2 ) n -O-phenyl-CH 2 -phenyl, -(CH 2 ) n - ⁇ henyl-(O-CH 2 - phenyl) 2 , -CH 2 -phenyl-C(O)-benzothiazole or a moiety of the formulae -(
- D is H, C,-C 6 lower alkyl, C,-C 6 lower aikoxy, -CF 3 or -(CH 2 ) n -CF 3 ;
- B and C are independently selected from phenyl, pyridinyl, pyrimidinyl, furyl, thienyl or pyrrolyl groups, each optionally substituted by from 1 to 3, preferably 1 to 2, substituents selected from H, halogen, -CN, -CHO, -CF 3 , -OH, -C,-C 6 alkyl, C,-C 6 aikoxy, -NH 2 or -NO 2 ; or a pharmaceutically acceptable salt thereof.
- the present invention also provides for a method of inhibiting the phospholipase enzyme activity of an enzyme, comprising administering to a mammalian subject a therapeutically effective amount of a compound of the present invention.
- Methods of treating an inflammatory response or condition comprising administering to a mammalian subject a therapeutically effective amount of a compound of the present invention are also provided.
- Pharmaceutical compositions comprising compounds of the present invention and a pharmaceutically acceptable carrier are also provided.
- Figs. 1-13 depict schemes for synthesis of compounds of the present invention. The depicted schemes are described in further detail below.
- aryl and substituted aryl are understood to include monocyclic, particularly including five- and six-membered monocyclic, aromatic and heteroaromatic ring moieties and bicyclic aromatic and heteroaromatic ring moieties, particularly including those having from 9 to 10 ring atoms.
- aryl groups are understood to be phenyl rings, including those found in phenoxy, benzyl, benzyloxy, biphenyl and other such moieties.
- the aryl and heteroaryl groups of this invention also include the following:
- a bicyclic ring moiety optionally containing from 1 to 3 ring heteroatoms selected from N, S or O including, but not limited to benzofuran, chromene, indole, isoindole, indoline, isoindoline, napthalene, purine, indolizine, indazole, quinoline, isoquinoline, quinolizine, quinazoline, cinnoline, phthalazine, or napthyridine.
- substituted aryl groups of this invention include such moieties being optionally substituted by from 1 to 3 substituents selected from halogen, C1-C10 alkyl, preferably C1-C6 alkyl, C1-C10 aikoxy, preferably C1-C6 aikoxy, -CHO, -COOH or esters thereof, -N02, -NH2, -CN, -CF3 or -OH or combinations thereof, such as -CH2CF3, -NH(CH3), etc.
- substituents selected from halogen, C1-C10 alkyl, preferably C1-C6 alkyl, C1-C10 aikoxy, preferably C1-C6 aikoxy, -CHO, -COOH or esters thereof, -N02, -NH2, -CN, -CF3 or -OH or combinations thereof, such as -CH2CF3, -NH(CH3), etc.
- a preferred subset of these groups include moieties formed from benzene, pyridine, napthylene or quinoline rings.
- a further preferred group includes those of furan, pyrrole, thiophene, pyrimidine, and morpholine rings.
- a preferred group of bicyclic aromatic groups includes benzofuran, indole, napthalene, and quinoline rings.
- alkyl, alkenyl and alkinyl groups referred to herein indicate such groups having from 1 to 10, preferably 1 to 6 carbon atoms, and may be straight, branched or cyclic. Unless indicated otherwise, it is preferred that these groups be straight or branched.
- Halogens herein are understood to include F, Cl, Br and I.
- Tables I- VI also report data for the listed compounds in the "LysoPC” assay and the Coumarine assay (see Example 88 below).
- assay results are reported as an "IQ” value, which is the concentration of a compound which inhibits 50% of the activity of the phospholipase enzyme in such assay.
- IQ is the concentration of a compound which inhibits 50% of the activity of the phospholipase enzyme in such assay.
- NA denotes that inhibitory activity was not detected from such compound in the corresponding assay and a blank box denotes that the compound was not tested in such assay as of the time of filing of the present application.
- phospholipase enzyme activity means positive activity in an assay for metabolism of phospholipids (preferably one of the assays described in Example 88 below).
- a compound has "phospholipase enzyme inhibiting activity” when it inhibits the activity of a phospholipase (preferably cPLA) in any available assay (preferably an assay described below in Example 88 or Example 89) for enzyme activity.
- a compound has (1) an I 0 value of less than about 25 ⁇ M, preferably less than about 6 ⁇ M, in the LysoPC assay; (2) an I 0 value of less than about 50 ⁇ M in the vesicle assay; (3) an I 0 value of less than about 1 ⁇ M in the PMN assay; (4) an IQ, value of less than about 15 ⁇ M in the Coumarine assay; and/or (5) measurable activity (preferably at least about 5 % reduction in edema, more preferably at least about 10% reduction, more preferably at least about 15% , most preferably about 20-30%) in the rat carrageenan-induced footpad edema test.
- Compounds of the present invention are useful for inhibiting phospholipase enzyme (preferably cPLA,) activity and, therefore, are useful in "treating” (i.e.. treating, preventing or ameliorating) inflammatory or inflammation-related responses or conditions (e.g. , rheumatoid arthritis, psoriasis, asthma, inflammatory bowel disease, and other diseases mediated by prostaglandins, leukotrienes or PAF) and other conditions, such as osteoporosis, colitis, myelogenous leukemia, diabetes, wasting and atherosclerosis.
- inflammatory or inflammation-related responses or conditions e.g. , rheumatoid arthritis, psoriasis, asthma, inflammatory bowel disease, and other diseases mediated by prostaglandins, leukotrienes or PAF
- the present invention encompasses both pharmaceutical compositions and therapeutic methods of treatment or use which employ compounds of the present invention.
- Compounds of the present invention may be used in a pharmaceutical composition when combined with a pharmaceutically acceptable carrier.
- a pharmaceutically acceptable carrier may also contain (in addition to a compound or compounds of the present invention and a carrier) diluents, fillers, salts, buffers, stabilizers, solubilizers, and other materials well known in the art.
- pharmaceutically acceptable means a non-toxic material that does not interfere with the effectiveness of the biological activity of the active ingredient(s).
- the characteristics of the carrier will depend on the route of administration.
- the pharmaceutical composition may further contain other anti-inflammatory agents. Such additional factors and/or agents may be included in the pharmaceutical composition to produce a synergistic effect with compounds of the present invention, or to minimize side effects caused by the compound of the present invention.
- the pharmaceutical composition of the invention may be in the form of a liposome in which compounds of the present invention are combined, in addition to other pharmaceutically acceptable carriers, with amphipathic agents such as lipids which exist in aggregated form as micelles, insoluble monolayers, liquid crystals, or lamellar layers in aqueous solution.
- Suitable lipids for liposomal formulation include, without limitation, monoglycerides, diglycerides, sulfatides, lysolecithin, phospholipids, saponin, bile acids, and the like. Preparation of such liposomal formulations is within the level of skill in the art, as disclosed, for example, in U.S. Patent No. 4,235,871; U.S. Patent No. 4,501,728; U.S. Patent No. 4,837,028; and U.S. Patent No. 4,737,323, all of which are incorporated herein by reference.
- the term "therapeutically effective amount” means the total amount of each active component of the pharmaceutical composition or method that is sufficient to show a meaningful patient benefit, i.e. , treatment, healing, prevention or amelioration of an inflammatory response or condition, or an increase in rate of treatment, healing, prevention or amelioration of such conditions.
- a meaningful patient benefit i.e. , treatment, healing, prevention or amelioration of an inflammatory response or condition, or an increase in rate of treatment, healing, prevention or amelioration of such conditions.
- the term refers to that ingredient alone.
- the term refers to combined amounts of the active ingredients that result in the therapeutic effect, whether administered in combination, serially or simultaneously.
- a therapeutically effective amount of a compound of the present invention is administered to a mammal having a condition to be treated.
- Compounds of the present invention may be administered in accordance with the method of the invention either alone or in combination with other therapies such as treatments employing other anti-inflammatory agents, cytokines, lymphokines or other hematopoietic factors.
- compounds of the present invention may be administered either simultaneously with the other anti- inflammatory agent(s), cytokine(s), lymphokine(s), other hematopoietic factor(s), thrombolytic or anti-thrombotic factors, or sequentially. If administered sequentially, the attending physician will decide on the appropriate sequence of administering compounds of the present invention in combination with other anti-inflammatory agent(s), cytokine(s), lymphokine(s), other hematopoietic factor(s), thrombolytic or anti-thrombotic factors.
- compositions of the present invention used in the pharmaceutical composition or to practice the method of the present invention can be carried out in a variety of conventional ways, such as oral ingestion, inhalation, or cutaneous, subcutaneous, or intravenous injection.
- compounds of the present invention will be in the form of a tablet, capsule, powder, solution or elixir.
- the pharmaceutical composition of the invention may additionally contain a solid carrier such as a gelatin or an adjuvant.
- the tablet, capsule, and powder contain from about 5 to 95 % compound of the present invention, and preferably from about 25 to 90% compound of the present invention.
- a liquid carrier such as water, petroleum, oils of animal or plant origin such as peanut oil, mineral oil, soybean oil, or sesame oil, or synthetic oils
- the liquid form of the pharmaceutical composition may further contain physiological saline solution, dextrose or other saccharide solution, or glycols such as ethylene glycol, propylene glycol or polyethylene glycol.
- the pharmaceutical composition contains from about 0.5 to 90% by weight of compound of the present invention, and preferably from about 1 to 50% compound of the present invention.
- compounds of the present invention When a therapeutically effective amount of compounds of the present invention is administered by intravenous, cutaneous or subcutaneous injection, compounds of the present invention will be in the form of a pyrogen-free, parenterally acceptable aqueous solution.
- parenterally acceptable protein solutions having due regard to pH, isotonicity, stability, and the like, is within the skill in the art.
- a preferred pharmaceutical composition for intravenous, cutaneous, or subcutaneous injection should contain, in addition to compounds of the present invention, an isotonic vehicle such as Sodium Chloride Injection,
- the pharmaceutical composition of the present invention may also contain stabilizers, preservatives, buffers, antioxidants, or other additives known to those of skill in the art.
- the amount of compound(s) of the present invention in the pharmaceutical composition of the present invention will depend upon the nature and severity of the condition being treated, and on the nature of prior treatments which the patient has undergone. Ultimately, the attending physician will decide the amount of compound of the present invention with which to treat each individual patient. Initially, the attending physician will administer low doses of compound of the present invention and observe the patient's response.
- the various pharmaceutical compositions used to practice the method of the present invention should contain about 0.1 ⁇ g to about 100 mg (preferably about .1 mg to about 50 mg, more preferably about lmg to about 2 mg) of compound of the present invention per kg body weight.
- the duration of intravenous therapy using the pharmaceutical composition of the present invention will vary, depending on the severity of the disease being treated and the condition and potential idiosyncratic response of each individual patient. It is contemplated that the duration of each application of the compounds of the present invention will be in the range of 12 to 24 hours of continuous intravenous administration. Ultimately the attending physician will decide on the appropriate duration of intravenous therapy using the pharmaceutical composition of the present invention.
- Compounds of the present invention can be prepared according to the following methods. Temperatures are in degrees Celsius.
- Indol-2-carboxylic acid ethyl ester I is converted to aldehyde II in two steps: reduction with lithium aluminum hydride (LAH) or other hydride in a suitable solvent such as tetrahydrofuran (THF) at 0°C, and then oxidation with an oxidizing reagent such as manganese dioxide in a solvent such as THF.
- LAH lithium aluminum hydride
- THF tetrahydrofuran
- an oxidizing reagent such as manganese dioxide in a solvent such as THF.
- Deprotonation of aldehyde II with a strong base such as potassium hexamethyldisilyl amide (KHMDS) in THF followed by reaction with a chloroformate in the presence of a base, such as triethyl amine, produces carbamate III.
- KHMDS potassium hexamethyldisilyl amide
- Ill is transformed into bromide IV in two steps: (1) reduction with sodium borohydride in an alcoholic solution and (2) reaction withcarbon tetrabromide in the presence of a phosphine reagent such as bis(diphenylphosphino)propane in dichloromethane.
- a phosphine reagent such as bis(diphenylphosphino)propane in dichloromethane.
- Displacement of the bromine in IV with potassium phenoxide, prepared by reaction of a phenol with KHMDS, in a suitable solvent such as THF or DMF affords ether V.
- V can be converted to either trifluoromethyl ketone VII or to carboxylic acid IX in different procedures.
- 2-Indolyl carboxylic acid ethyl ester I is deprotonated with a strong base such as sodium hydride (NaH) in THF, and then reacted with a suitable alkyl bromide to give X.
- a strong base such as sodium hydride (NaH) in THF
- a suitable alkyl bromide to give X.
- Hydrolysis of X with a aqueous base such as sodium hydroxide and reaction with aniline or a substituted aniline in the presence of a carbodiimide such as dimethylaminopropyl ethylcarbodiimide hydrochloride (EDCI) in a suitable solvent such as dichloromethane affords amide XL XI is hydrolyzed to corresponding acid XII in a aqueous base such as sodium hydroxide.
- a carbodiimide such as dimethylaminopropyl ethylcarbodiimide hydrochlor
- Indole I can be brominated on the 3-position by reaction with a bromine or N- bromosuccinimide in a suitable solvent such ascarbon tetrachloride or dichloromethane to yield bromide XIII.
- a suitable solvent such ascarbon tetrachloride or dichloromethane
- Reaction of XIII with a suitable alkyl bromide in the presence of a strong base such as NaH in THF or DMF affords indole XIV.
- Palladium mediated coupling of XIV with a suitable alkene in the presence of phosphine and a base such as triethyl amine produces 3-substituted indole XV.
- XV can be converted to amide XVII in two step reactions:
- Indole I can be converted to XXI in two steps: (1) reduction with LAH in a solvent such as THF and (2) silylation with t-butyldimethylsilyl chloride (TBDMSC1) in a solvent such as dichloromethane or DMF in the presence of a base such as imidazole.
- a solvent such as THF
- TBDMSC1 t-butyldimethylsilyl chloride
- the silyl group on XXII is removed using tetrabutylammonium fluoride in a solvent such THF, the resulting alcohol is then converted to bromide using carbon tetrabromide and bis(diphenylphosphino)ethane in a solvent such as dichloromethane to yield bromide XXIII.
- Aldehyde II prepared by Method A, can be alkylated by a suitable alkyl bromide (or iodide), such as benzyl bromide or ethyl iodide in the presence of a strong base such as sodium hydride or KHMDS in a solvent such as DMF to yield XXV.
- XXV can be converted to an unsaturated acid XXVI by two steps: (1) Wittig reaction with a suitable reagent such as trimethyl phosphonoacetate in the presence of a base such as sodium hydride in a solvent such as THF and (2) Hydrolysis by aqueous sodium hydroxide.
- Indole I is reduced with LAH in a solvent such as THF.
- BOC t-butoxycarbonyl
- (BOQO) di-t-butyldicarbonate
- the hydroxyl group in XXIX is mesylated using mesyl chloride and triethylamine in a solvent such as dichloromethane, and then displaced by either a thiol or an alcohol as described in METHOD D to produce indoline XXX.
- XXXI Deprotection of XXX using trifluoroacetic acid affords XXXI, which is either acylated (acyl chloride, triethylamine, dichloromethane) or alkylated (alkyl halide, K : C0 3 , DMF) to afford XXXII, or XXXIII respectively.
- Carboxylic acid XXXIV is converted to aldehyde XXXV in two steps: (1) reaction with N,0- dimethylhydroxy amine in the presence of EDCI in a solvent such as dichloromethane, and
- DIBAL diisobutyl aluminum hydride
- METHOD H Aldehyde XXXV prepared in METHOD G, is subjected to a Wittig reaction using methyl triphenylphosphonium iodide in the presence of a strong base such as KHMDS or NaH in a solvent such as THF to afford alkene XL.
- a strong base such as KHMDS or NaH in a solvent such as THF
- Reduction of the nitro group of XL with iron powder in an ammonium chloride solution, followed by treatment with benzyl chloroformate in the presence of a base such as triethyl amine produces carbamate XLI.
- XLI is treated with iodine in a basic solution such as aqueous NaHCQ in THF to yield iodide XLII.
- Displacement of the iodine on XLII with lithium benzoate in a solvent such as DMF, followed by hydrolysis with NaOH affords alcohol XLIII.
- Indoline XXVIII prepared in METHOD F or METHOD H. can be either acylated by reaction with an acyl chloride in the presence of a base such as triethyl amine or alkylated using alkyl halide in the presence of KC0 3 in a solvent such as DMF to produce alcohol
- XL VII can be alkylated on the amide nitrogen by treatment with alkyl halide and strong base such as NaH in DMF. Hydrolysis of the resulting amide with aqueous base such as NaOH gives acid XLIX. XLIV can also be directly hydrolyzed with NaOH to a carboxylic acid XLVIII.
- Ester L can be deprotonated with a strong base such as lithium diisobutylamide (LDA) in a solvent such as THF, and subsequently alkylated with an alkyl halide such as methyl iodide to give LI.
- LDA lithium diisobutylamide
- Reduction of LI to amine LIII can be accomplished using hydrogenation catalyzed by palladium in a solvent such as ethanol.
- L can be oxidized to alcohol LH using LDA and oxaziridine in a solvent such as THF.
- Alkylation of LII with a alkylating reagent such as methyl iodide in the presence of a strong base such as NaH in DMF, followed by catalytic hydrogenation in the presence of palladium produces amine LIV.
- METHOD K illustrates the synthesis of substituted aminobenzoic acid esters.
- Mono-acid LV can be converted to amide LVI by the following steps: (1) reaction with oxalyl chloride in dichloromethane to form acid chloride and (2) treatment with a suitable amine such as dimethyl amine. Reduction of the nitro group to the amine is accomplished with hydrogenation catalyzed by palladium as described in METHOD J. LV can be reduced to alcohol LVIII with hydroborane-THF complex in THF. Protection of the hydroxy group as a silyl ether using TBDMSC1 in the presence of imidazole and subsequently, reduction of the nitro group (H 2 / Pd-C) to the amine affords LIX.
- LVIII can be convened to the secondary alcohol LX in two steps: (1) oxidation with a suitable reagent such as manganese dioxide (Mn0 2 ) in ethyl acetate and (2) addition of a desired Grignard reagent such as methyl magnesium bromide in THF. Oxidation of LX with MnQ in THF and reduction of the nitro group (H 2 / Pd-C) produces ketone LXIII. Reduction of LVII (H / Pd-C) yields LXI.
- a suitable reagent such as manganese dioxide (Mn0 2 ) in ethyl acetate
- Grignard reagent such as methyl magnesium bromide
- Alcohol LXIV prepared in METHOD I, can be debenzylated by hydrogenolysis catalyzed by palladium on carbon in a solvent such as ethanol. The resulting alcohol is treated with p- methoxybenzyl chloride in the presence of KC0 3 in a solvent such as THF to afford LXV. Alcohol LXV can be transformed into ether or sulfide LXVI by the procedures described in METHOD D.
- Step 1 2-C5-Phenylmethoxy)indolyl aldehyde
- Step 2 Benzyl (l-(2-fo ⁇ nyl-5-phenylmethoxy)indolyl)formate
- Step 3 Benzyl ⁇ -(2-hydroxymethyl-5-phenylmethoxy1indolyl)formate
- Step 4 Benzyl ⁇ -f2-bromomethyl-5-phenylmethoxy)indolyl)formate
- Step 5 Benzyl Cl-(2-('2-formylphenoxy)methyl-5-phenylmethoxy indolyl)formate
- Step 6 120 mg (0.24 mmol) of the aldehyde of step 5 was dissolved in 11 mL of 5: 1 :5 THF- acetonitrile-2,2-dimethylethanol. To this solution was added a solution of 56 mg (0.5 mmol) of sodium chlorite in 0.5 mL water and 1 drop of aqueoues hydrogen peroxide solution. After 4 hours, another 56 mg (0.5 mmol) of sodium chlorite was added. The mixture was stirred at room temperature for three days. Aqueous work up and flash chromatography using 2.5: 1:0.05 hexane:ethyl acetate-acteic acid afforded 110 mg of the title compound.
- Step 1 Benzyl (l-f2-(2-(l-hydroxy-2.2.2-trifluoroethyl phenoxy)methyl-5- phenylmethoxy ndolyP-formate
- Step 1 Ethyl 2-Cl-benzyl-5-benzyloxy)indolecarboxylate
- Step 3 Ethyl 3-(2-( l-benzyl-5-benzyloxy)indolecarboxamido)benzoate
- step 2 l-(3-dimethylaminopropyl)-3- ethylcarbodiimide (EDCI) (0.32 g, 1.66 mmol), 4-dimethylaminopyridine (DMAP) (0.018 g, 0.15 mmol) and ethyl 3-aminobenzoate (0.27 g, 1.66 mmol) were stirred in tetrahydrofuran (9 mL) at room temperature overnight. The next day the reaction was diluted with ethyl acetate and water, extracted with ethyl acetate (3X), dried over magnesium sulfate and concentrated.
- EDCI l-(3-dimethylaminopropyl)-3- ethylcarbodiimide
- DMAP 4-dimethylaminopyridine
- ethyl 3-aminobenzoate 0.27 g, 1.66 mmol
- Ethyl 5-methoxy-2-indolcarboxylate (30 g, 102 mmol) is dissolved in 250 mL of THF and cooled to P C and Lithium Aluminum Hydride (LAH) (255 mL of a 1.0 M solution in THF) is added via addition funnel over 40 minutes. The reaction was stirred a further 2 hours at 0° C and then worked up by the addition of 4N NaOH (190 mL). The resulting salts are filtered and washed with ethyl acetate (3X400 mL), the filtrates are combined and dried over MgS0 and concentrated to yield 24.8 g of alcohol, which was used for the next reaction directly.
- LAH Lithium Aluminum Hydride
- Step 2 2-(5-methoxy)indolylmethoxy-tert-buthyldimethylsilane
- DMF dimethyl methoxy-buthyldimethylsilane
- imidazole 5.5g, 81.5 mmol
- t- butyldimethylsilyl chloride 5.4g, 35.8 mmol
- the reaction was poured into water and extracted with ethyl acetate (3X). Organic layers were dried over magnesium sulfate and concentrated.
- Step 3 3-(2-tert-butydimethylsilyloxymethyl-5-methoxy ndolyl C2.4-bisd . l- dimethypropyl)phenoxy)methyl ketone
- Step 4 S- ⁇ -tert-butydimethylsilyloxymethyl-S-methoxy-l-methyOindolyl (2.4-bisd . l- dimethypropyl ' )phenoxy)methyl ketone
- Step 5 3-C2-Hydroxymethyl-5-methoxy-l-methyl)indolyl (bis-2,4- .1. dimethy lpropyPphenoxylmethyl ketone
- Step 6 Methyl 3-f2-(3-f2.4-bisd . l-dimethypropyl)phenoxy)acetyl-5-methoxy-l- methylindolyl)methylthioacetamido)-4-methoxybenzoate
- EXAMPLES 13. 14. 15 and .16 in Table I were prepared by the procedures of Example 12 using Ethyl 2-(5-benzyloxy)indolecarboxylate, acetyl chlorides and suitable alkyl halides.
- Ethyl 5-benzyloxy-2-indolecarboxylate (30 g, 102 mmol) was dissolved in 250 mL of THF and cooled to 0° C, to which Lithium Aluminum Hydride (LAH) (255 mL of a 1.0 M solution in THF) was added via addition funnel over 40 minutes. The reaction was stirred a for 2 hours at 0 °C and then worked up by the addition of 4N NaOH (190 mL). The resulting salts were filtered and washed with ethyl acetate (3X400 mL), the filtrates were combined, dried over MgS0 and concentrated to yield 24.8 g.
- LAH Lithium Aluminum Hydride
- Step 2 tert-Butyl l-(5-benzyloxy-2-hydroxymethy)lindolinylformate 25 g (85 mmol) of crude alcohol, prepared in step 1, and 4-dimethylamino pyridine (DMAP) (1.19 g, 9.78 mmol) were dissolved in dichloromethane (180 mL). The solution was cooled to CP C and then triethylamine (13.6 mL, 98 mmol) was added to it. After 10 minutes of stirring a solution of di-tert-butyl dicarbonate (21.3 mL, 98mmol) dissolved in dichloromethane (20 mL) was added via syringe pump over 2 hours.
- DMAP 4-dimethylamino pyridine
- the carbamate, prepared in step 2 (15.25 g, 43 mmol) was dissolved in dichloromethane (180 mL) and treated with triethylamine (9.0 mL, 64.4 mmol). The solution was cooled to -10 p C at which time mesyl chloride (4.3 mL. 56 mmol) was added over 5 minutes. The reaction was stirred for a further 2 hour at -10°C, it was then concentrated and used directly for the next displacement reaction.
- Step 5 Ethyl 3-f2-f5-henzyloxy-l-tert- butoxycarbonvPindolinyPmethylthioacetamidobenzoate
- Step 7 Ethyl 3-(2-(5-benzyloxy- 1 -(2 ,4-bisf 1.1 -dimethy PpropyDphenoxyacetyPindolinvD methylthioacetamidobenzoate
- step 7 The ester (0.231 g, 0.31 mmol) of step 7 was dissolved in THF (4.3 mL), methanol
- Step 3 2-(5-Benzyloxy-l-f2.4-bisd . l- dimethy propyPphenoxyacetyPindolinylmethylthioacetic acid
- Step 4 Methyl 3-(2-(5-benzyloxy- 1 -(2.4-bisf 1.1 -dimethy)propyPphenoxyacetyPindolinyl) methylthioacetamido-4-methylbenzoate
- the titled compound was prepared from ester, prepared in step 4, according to the procedure described in step 3.
- Step 1 2-(5-Benzyloxy-l- .5-bisCtrifluoromethyPphenoxyacetyl)indolinyPmethanol
- Step 2 Ethyl 2-(5-benzyloxy-l-(3.5-bis(trifluoromethyPphenoxyacetyPindolinyl) methylthioacetate
- Step 1 5-(2-(-5-Benzyloxy-l-G.5-bisCtrifluoromethyl)phenoxyacetyPindolinyl) methy lthioacetamido)benzene- 1.3-dicarboxylate
- Step 1 Methyl 5-('2-C-5-benzyloxy-l-(3.5-bis(trifluoromethyl)phenoxyacetyl)indolinyl) methylthioacetamido)-3-tert-butyldimethylsilyloxymethylbenzoate
- This compound was prepared according to the procedure described in step 1 of Example 38.
- Step 2 Methyl 5-(2-C-5-benzyloxy-l-C3.5-bis(trifluoromethyPphenoxyacetyPindolinyl) methylthioacetamido)-3-hydroxymethylbenzoate
- the titled compound was prepared according to the procedure described in step 2 of Example 38.
- EXAMPLE 42 in table 3 was prepared according to the procedures described in Example 41.
- Step 1 2-('5-Hydroxy-l-C3.5-bis(trifluoromethvPphenoxyacetyPindolinyPmethanol
- Step 2 2-(5-C4-Methoxy)henzyloxy-l-f3.5- bisftrifluoromethyPphenoxyacetyPindolinvPmethanol
- Step 3 Methyl 5-(2- ⁇ '-5-(4-methoxy)benzyloxy-l-(3.5-bis(trifluoromethyPphenoxyacetyP indolinvPmethylthioacetamido)benzene-1.3-dicarboxylate
- Step 4 Methyl 5-(2- ⁇ 5-Hydro ⁇ y-l-(3.5-bisftrifluoromethyPphenoxyacetyP indolinyPmethylthioacetamido)benzene- 1.3-dicarboxylate
- Stepl Methyl 5-(2-C5-(3.5-Dibromo)benzyloxy-l-(3.5-bis(trifluoromethyPphenoxyacetyP indolinyPmethylthioacetamidolbenzene- 1.3-dicarboxylate
- the titled compound was prepared from the ester, prepared in step 1 , according to the procedure described in step 5 of Example 44.
- Example 44 but using corresponding alkylating reagent.
- This compound was prepared according to the procedures described in step 6 of Example 17. but with methyl 4-methoxybenzoate.
- Step 2 Methyl 3-(2-(5-benzyloxy-l-C2-naphthoxyacetvPindolinyPmethylthioacetamido)-4- methoxybenzoate
- Step 1 Ethyl 3-C2-( " 5-benzyloxy-l-tert-butoxycarbonyl)indolinyl)methylsulfonyl acetamidobenzoate
- the titled compound was prepared according to the procedure described in step 3 of Example 59.
- Step 1 5-Benzyloxy-l-(2.4-bisd . l-dimethy)propyPphenoxyacetyl)-2-hydroxymethylindoline
- Step 2 2-(5-Benzyloxy- 1 -C2 ,4-bisf 1.1 -dimethy)propyl)phenoxyacetyPindolinylmethyl _ methylsulfonate
- Step 3 Methyl 2-(2-(-5-benzyloxy-l-C2.4-bisd . l-dimethy ' )propyPphenoxyacetyP indolinyPmethylthiobenzoate
- the titled compound was prepared according to the procedure described in step 3 of Example 59.
- EXAMPLE 68 was prepared according to the procedures described in Example 67.
- EXAMPLE 71 was prepared according to the procedures described in Example 70. but using allyl bromide.
- Step 1 Ethyl 3-(2-(5-benzyloxy-1 -C2-C4- pyridinyPethyPindolinyPmethylthioacetamidobenzoate
- the titled compound was prepared according to the procedure described in step 3 of Example 59.
- Step 1 Ethyl 3-( ' 2-(5-benzyloxy-l-('2-naphthyPmethy)indolinyPmethylthioacetamidobenzoate
- the titled compound was prepared according to the procedure described in step 3 of Example 59.
- Step 1 2-C2-( " -5-Benzyloxy- 1 -d .1 -dimethyPethoxycarbonyPindoliny Pmethyl methylsulfonate
- tert-Butyl l-(5-benzyloxy-2-hydroxymethy)lindolinylformate (6.72 g, 19 mmol), prepared in step 2 of Example 17. was dissolved in CHC1 2 (80 mL, dried over MgS0 4 before use). The clear yellow solution was cooled in a dry-ice bath. EjN (4.0 mL) was then added followed by methanesulfonyl chloride (2.0 mL). The reaction mixmre was stirred for
- Step 2 Methyl 2-(2-(5-Benzyloxy-l-d . l-dimethyl)ethoxycarbonyl)indolinyPmethylthio benzoate
- Mesylate (7.2 g, 1.8 mmol), prepared in step 1, was dissolved in DMF (50 mL). The clear light brown solution was degassed by vigorously bubbling with Ar for 30 min. Cesium carbonate (13.8 g) was added followed by methyl thiosalicylate (2.4 mL). The solution changed to a bright yellow and the suspension was stirred overnight. Methyl thiosalicylate (0.15 mL) was added to complete the reaction and the mixmre was stirred overnight.
- ester (1 g), prepared in step 3 was dissolved in DMF (6 mL).
- p-Benzylbenzyl bromide was added (1 eq) followed by KC0 3 (1 eq).
- the reaction mixture was stirred overnight at room temperature.
- additional p-benzylbenzyl bromide (0.5 eq) was added and the reaction was stirred for another 2 hours.
- the reaction was diluted with HO and extracted with EtOAc (2 x). The organic layers were combined and dried over MgSQ.
- the MgS0 4 was filtered and the solvent was evaporated to give an oily material which was dried overnight on high vacuum to give the product (1.59 g, 109 % yield).
- Step 1 Methyl l-(5-Benzyloxy-2-(hydroxymethyPindolinyPmethylbenzoate
- Step 2 Methyl 4-d-(5-Benzyloxy-2-0 is-2.4-trifluoromethyPbenzyloxymethyPindolinyl) methylbenzoate
- the titled compound was prepared according to the prodedure described in step 5 of
- Step 1 2-d-(2.4-Bis(trifluoromethyPbenzyl)indolinyPcarboxylic acid
- 2-Indolinylcarboxylic acid (0.43 g, 2.6 mmol) was dissolved in DMF (5 mL), placed under N 2 , and cooled to Of C, the sodium hydride (0.26 g of a 60 % dispersion, 6.5 mmol) was added and stirring was continued for 1 hour at this temperamre.
- 2,4- Bis(trifluoromethyl)benzyl bromide (1.22 mL, 6.5 mmol) was next added and the reaction was warmed to room temperamre overnight.
- the reaction was then diluted with 1/2 samrated ammonium chloride/ethyl acetate, the aqueous layer was extracted with ethyl acetate (3X), the organic layers were dried over magnesium sulfate and concentrated.
- the crude product was purified via chromatography (hexane:ethyl acetate 9: 1) to yield 0.96 g of the ester.
- the resulting ester (0.87 g, 0.1.41 mmol) was dissolved in THF/ methanol and then
- the titled compound was prepared according to the prodedure described in Example 84, but using phenylsulfonylamide.
- Step 1 2-TrimethyIsilylethyl l-(5-benzyloxy-2-hydroxymethyPindolinylformate
- Step 3 2-Trimethylsilylethyl l-C5-f4-methoxy)benzyloxy-2-hydroxymethyl)indolinylformate
- Step 4 2-Trimethylsilylethyl l-CS- ⁇ -methoxy ⁇ enzyloxy ⁇ -bromomethyPindolinylformate
- Step 5 2-Trimethylsilylethyl l-(5-(4-methoxy)benzyloxy-2-azidomethyPindolinylformate
- Step 6 2-Trimethylsilylethyl l-(5-(4-methoxy)benzyloxy-2-aminomethyPindolinylformate
- Step 7 Methyl 5-(2-(5-Methoxybenzyloxy-l-(2-trimethylsilyloxy)ethoxycarbony)lindolinyP methylaminocarboxamido- 1.3-benzenedicarboxylate
- the product was extracted with ethyl acetate, and the combined organic layers were washed with water, samrated aqueous NaHCQ, brine and dried over MgS0 4 .
- the crude product was purified by flash chromatography using 10% MeOH/CHCl 2 to afford 0.78 g of the product.
- Step 8 Methyl 5-(2-(5-Methoxybenzyloxy)indolinyl)methylaminocarboxamido-1.3- benzenedicarboxylate
- Step 9 Methyl 5-(2-(5-Methoxybenzyloxy-l-(bis-2.4-trifluoromethyPbenzyPindolinyP methylaminocarboxarnido- 1.3-benzenedicarboxylate
- EXAMPLE 87 was prepared according to the prodedure described in Example 86. but using 4-(3,5-bis(trifluoromethyl)phenoxymethyl)benzyl bromide.
- Step 1 Bisfmethyl 4-methoxy-3-dithioacetamidobenzoate
- reaction mixmre was allowed to warm to -30°C over 20 min. and then cooled to -50 °C again.
- the reaction mixture was quenched with HO (500 mL) at -50 °C and warmed up to room temperamre and stirred for 0.5 h.
- the reaction mixture was partitioned between CH 2 C1 2 (500 mL) and H 2 0.
- the aqueous layer was extracted with CHC1 2 (3 x 500 mL).
- the combined CH,C1 2 extracts were concentrated in vacuo to give a yellow oil. This was added slowly to a 2 M solution of NaOH (2 L) cooled at 0°C and stirred at room temperature overnight.
- the titled compound was prepared from nitro compound of step 1 according to the procedure described in step 4 of Intermediate 3.
- the titled compound was prepared from nitro compound, prepared in step 1 of Intermediate 3, according to the procedure described in step 4 of Intermediate 3.
- Step 1 Methyl 2-(3-nitro-4-methoxyphenyl)-2-methylacetate
- the titled compound was prepared from nitro compound, prepared in step 1 , according tothe procedure described in step 4 of Intermediate 3.
- Step 1 Methyl 2-(3-nitro-4-methoxyphenyl)-2-allylacetate
- This compound was synthesized form ester, prepared in step 1 of Intermediate 3, according to the procedure described in step 1 of Intermediate 6, but using allyl bromide.
- the titled compound was prepared from 2-naphthol according to the procedure described in of Intermediate 8.
- the titled compound was prepared from 3,5-bis(trifluoromethyl)phenol according to the procedure described in of Intermediate 8.
- the titled compound was prepared from nitro compound, prepared in step 1, according to the procedure described in step 4 of Intermediate 3.
- the titled compound was prepared from nitro compound, prepared in step 1 , according to the procedure described in step 4 of Intermediate 3.
- Step l Methyl 5-nitro-3-d -hydro xy)ethylbenzoate
- the titled compound was prepared from nitro compound, prepared in step 2, according to the procedure described in step 4 of Intermediate 3.
- Step 1 Bis 2-bromoethyPdisulfide
- Step 2 Bis-Onethyl 4-methoxy-3-(2-dithioethyPaminobenzoate Bromide (0.39 mg, 1.387 mmol), prepared in step 1, and methyl 3-amino-4-methoxy benzoate (1.00 g, 5.51 mmol) were added into a flask, flush with nitrogen and take up in DMF (5 mL) and then heat to 60° C for 24 hours at which time the reaction was diluted with ethyl acetate and quenched into water, extracted with ethyl acetate (3X), the combined organic layers were washed with water (3X), dried and concentrated to yield 1.27 g of a product that was purified by chromatography (hexane:ethyl acetate 5: 1 to 3: 1) to yield 0.15 g of the desired product.
- the aldehyde is reacted with the alpha-carbon of a heterocycle such at 2,4-thiazolidinedione or rhodanine or 2-thiohydantoin in the presence of a base such a potassium carbonate or potassium hydroxide in a solvent system such a wate ⁇ efhanol or ethanol.
- a base such as potassium carbonate or potassium hydroxide
- a solvent system such as a wate ⁇ efhanol or ethanol.
- the final acid may then be realized by cleavage of the ester with hydrogen fluoride in a solvent such as acetonitrile.
- Indole-2-carboxylic acid was alkylated with an appropriate alkyl bromide which was then subjected to Suzuki coupling conditions using Pd(PPh 3 ) as a catalyst in a mixed solvent (ethanol-benzene- water) at elevated temperature to give the l-alkyl-5-substiruted indole.
- the starting material for the inhibitors in this class 2-Ethoxycarbonyl-5-benzyloxyindole I, was deprotonated with a suitable base such as sodium hydride and alkylated on the nitrogen atom with selected electrophiles such as alkyl or benzyl halides to provide compounds II. Saponification of the ester functionality with a base such as aqueous sodium hydroxide in miscible solvents such as tetrahydrofuran and methanol gave inhibitors III.
- a suitable base such as sodium hydride and alkylated on the nitrogen atom with selected electrophiles such as alkyl or benzyl halides
- Acid isosteres such as tetrazole were prepared from the carboxylic acids I via the nitriles III . Conversion to the nitriles was accomplished through primary amide formation of the acid functionality via the acid chloride with a suitable reagent such as oxalyl chloride and reaction with ammonia followed by a dehydration sequence using a suitable reagent such as oxalyl chloride and a base such as pyridine.
- the nitriles such as UI could be converted to the tetrazoles by reaction with an azide source such as sodium azide in an appropriate high boiling point solvent such as N-methyl pyrrolidinone to give compounds such as IV.
- 2-Indolyl carboxylic acid ethyl ester I is deprotonated with a strong base such as sodium hydride (NaH) in THF, and then reacted with a suitable alkyl bromide to give VI.
- a strong base such as sodium hydride (NaH) in THF
- a suitable alkyl bromide to give VI.
- Hydrolysis of VI witha aqueous base such as sodium hydroxide and reaction with aniline or a substituted aniline in the presence of a carbodiimide such as dimethylaminopropylethyl carbodiimide hydrochloride (EDCI) in a suitable solvent such as dichloromethane affords amide VII.
- Amide VII is hydrolyzed to corresponding acid VIII in a aqueous base such as sodium hydroxide.
- Aldehyde K is prepared from Indol-2-carboxylic acid ethyl ester I in two steps: (1) Reduction with lithium aluminium hydride or other hydride in a suitable solvent such as THF at 0°C and (2) oxidation with an oxidizing reagent such as manganese dioxide in a solvent such as THF.
- Aldehyde LX can be alkylated by a suitable alkyl bromide (or iodide), such as benzyl bromide or ethyl iodide in the presence of a strong base such as sodium hydride or KHMDS in a solvent such as DMF to yield indole X .
- Indole X can be converted to an unsaturated acid XI in two steps: (1) Wittig reaction with a suitable reagent such as trimethyl phosphonoacetate in the presence of a base such as sodium hydride in a solvent such as THF and (2) Hydrolysis by aqueous sodium hydroxide.
- Indole I can be converted to II in two steps: (1) reduction with LAH in a solvent such as THF and (2) silylation with t-butyldimethylsilyl chloride (TBDMSC1) in a solvent such as dichloromethane or DMF in the presence of a base such as imidazole.
- TBDMSC1 t-butyldimethylsilyl chloride
- a base such as imidazole.
- Grignard reagent such as ethyl magnesium bromide
- a suitable acyl chloride such as acetyl chloride in ether
- alkylation on the nitrogen with an alkyl halide such as ethyl bromide in the presence of a strong base such as NaH in DMF affords ketone III.
- the silyl group on III is removed using tetrabutylammonium fluoride in a solvent such as THF, the resulting alcohol is then converted to bromide using carbon tetrabromide and bis(diphenylphosphino)ethane in a solvent such as dichloromethane to yield bromide IV.
- R" halogen, CN, alkyl, aikoxy, alkoxycarbonyl, amido, acyl, H, OH
- R aikoxy, benzyloxy, phenoxy, halogen, CN, N0 2 , alkyl or aryl
- R' alkyl, aryl
- Step 1 The aldehyde from Example 124, (5.2 g) was suspended in ethanol (150 mL). To the thick slurry was added 2,4-thiazolidinedione (1.28g) and potassium carbonate (6.1g). The mixture was heated in a bath at 60 °C (later dropped to 45 °C). After 1 h TLC showed no reaction. Sodium hydroxide (2.1 g) was added and the mixture was heated at 58 °C. After 45 minutes the TLC showed reaction progress. Additional 2,4-thiazolidinedione (0.1 g) was added. The mixture was stirred overnight at room temperature.
- Step 2 To the material prepared in step 1 (1.1 g) in DMF (15 mL) at 0 °C was added sodium hydride (0.08 g. 60% dispersion in mineral oil). The suspension was stirred for 30 minutes. To the reaction mixture was added the benzyl bromide (0.54 g) and the reaction was stirred overnight. Water was added and the mixture was extracted with ethyl acetate. The combined organic layers were concentrated. Column chromatography (1:6 ethyl acetate: hexane to 1:4 ethyl acetate:hexane) afforded the desired product (1.18 g, 75%) as a yellow solid.
- Step 3 To the material prepared in step 2 (0.34 g) in acetonitrile (15 mL) was added HF (48% aqueous, 3.7 mL) via syringe. The reaction was stirred overnight. The reaction was not complete by TLC therefore THF was added to dissolve the starting material and additional HF (0.6 mL) was added. The reaction was stirred for 2 h after which the TLC showed reaction completion. Water was added which resulted in the formation of a yellow solid. The yellow solid was dissolved in ethyl acetate, washed with brine, dried over MgSO 4 and concentrated. The resulting crude solid was suspended in ethanol and stirred for 30 min, filtered and dried to afford the title compound (140 mg, 48%) as a yellow solid.
- Step 1 - The desired intermediate was prepared as illustrated in Example 88, step 1, starting with the appropriate indole.
- Step 2 - The desired intermediate was prepared from the above intermediate as illustrated in Example 88, step 2, using the appropriate alkylating agent.
- Step 3 The title compound was prepared from the above intermediate as illustrated in Example 88, step 3.
- Step 1 - The desired intermediate was prepared as illustrated in Example 88, step 1, starting with the appropriate indole.
- Step 2 - The desired intermediate was prepared from the above intermediate as illustrated in Example 88, step 2, using the appropriate alkylating agent.
- Step 3 The title compound was prepared from the above intermediate as illustrated in Example 88, step 3.
- Step 2 - The desired intermediate was prepared from the above intermediate as illustrated in Example 88, step 2, using the appropriate alkylating agent.
- Step 3 The title compound was prepared from the above intermediate as illustrated in Example 88, step 3.
- Step 1 - The desired intermediate was prepared as illustrated in Example 88, step 1, starting with the appropriate indole.
- Step 2 - The desired intermediate was prepared from the above intermediate as illustrated in Example 88, step 2, using the appropriate alkylating agent.
- Step 3 The title compound was prepared from the above intermediate as illustrated in Example 88, step 3.
- Step 1 - The desired intermediate was prepared as illustrated in Example 88, step 1, starting with the appropriate indole.
- Step 2 - The desired intermediate was prepared from the above intermediate as illustrated in Example 88, step 2, using the appropriate alkylating agent.
- Step 3 The title compound was prepared from the above intermediate as illustrated in Example 88, step 3.
- the compounds of the following Examples 101-106 were prepared as illustrated in Example 88, step 1, starting with the appropriate indole and rhodanine.
- Step 1 The desired intermediate was prepared as illustrated in Example 88, step 1, starting with the appropriate indole and rhodanine.
- Step 2 - The desired intermediate was prepared from the above intermediate as illustrated in Example 88, step 2, using the appropriate alkylating agent.
- Step 3 The title compound was prepared from the above intermediate as illustrated in Example 88, step 3.
- Example 117A To a suspension of the acid prepared in Example 117A (1.5g, 3.0mmol) in CH 2 C1 2 (20ml) was added oxalyl chloride (0.8ml, 9.1mmol) and three drops of DMF. The mixture became homogeneous and was stirred for lh at rt. The reaction was concentrated and redissolved in CH 2 C1 2 (5ml) and NH 4 OH (2.0ml) was added. The biphasic mixture was stirred for 24h and concentrated. The remaining aqueous residue was extracted with CH 2 C1, and the combined organic layers washed with brine, dried and concentrated to give 1.4g (95%) of the desired intermediate as a yellow powder.
- EXAMPLE 1 19 benzyl 1 -(4- ⁇ [3 ,5-bis(trifluoromethyl)phenoxy]methyl ⁇ benzyl)-2-( 1 H- 1 ,2,3 ,4-tetraazol-5-yl)- 1 H- indol-5-yl ether acid was prepared in an analogous manner to Example 118 according to steps 1-3 starting from the acid prepared in EXAMPLE 117C.
- Example 101 The thiasolidinedione prepared in Example 101 (O.lg, 0.2mmol), was alkylated by treatment with sodium hydride (0.006g, 0.22mmol), and the bromomethyl SEM ester (0.058g, 0.2mmol) in
- Step 1 To ethyl 5-benzyloxy-2-indolcarboxylate (1 g, 3.4 mmol) in 12 ml of DMF, sodium hydride (0.163g, 60% oil dispersion, 4.07 mmol) is added at room temperature. The reaction is stirred for 30 minutes. a-Bromo-a'-[3,5-bis(trifluoromethyl)phenoxyl]-p-xylene (1.54 g, 3.73 mmol) is added at this time and the reaction stirred overnight. On completion of the reaction (monitored by TLC) it is quenched with water, extracted with ethyl acetate (3X). Organic layers are dried over magnesium sulfate, concentrated and used for the next step.
- sodium hydride 0.163g, 60% oil dispersion, 4.07 mmol
- a-Bromo-a'-[3,5-bis(trifluoromethyl)phenoxyl]-p-xylene (1.54 g, 3.73 m
- Step 2 The ester ( 2.1 g, 3.39 mmol) is dissolved in 40 mL of 1/1 THF/ methanol and then IN sodium hydroxide (15 mL) is added and the resulting mixture is stirred for 16 hours at RT, workup gave crude product that is purified via chromatography (1:1 Hexane:Ethyl acetate with 1% acetic acid) to yield (1.73 g, 85%) of solid.
- EXAMPLE 123 5-((ri-benzyl-5-(benzyloxy)-lH-indol-2-yllcarbonyI ⁇ amino)isophthalic acid
- Step 1 This intermediate was prepared according to the procedure described in Example 122, but using benzyl bromide.
- Step 2 The acid (0.27 g, 0.75 mmol) prepared in step 1, EDCI (0.18 g, 0.97 mmol), DMAP (3 mg, 0.02 mmol) and dimethyl-5 aminoisophthalate (0.18g, 0.75 mmol) were dissolved in THF
- Step 3 The title compound was prepared from ester, prepared in step 2 above, according to the procedure described in step 2, Example 122.
- Step 1 Ethyl 5-benzyloxy-2-indolcarboxylate (30 g, 102 mmol) is dissolved in 250 mL of THF and cooled to 0° C and Lithium Aluminum Hydride (LAH) (255 mL of a 1.0 M solution in THF) is added via addition funnel over 40 minutes. The reaction was stirred a further 2 hours at 0° C and then worked up by the addition of 4N NaOH (190 mL). The resulting salts are filtered and washed with ethyl acetate (3X400 mL), the filtrates are combined and dried over MgSO 4 and concentrated to yield 24.8 g (96%).
- LAH Lithium Aluminum Hydride
- Step 2 Indole alcohol (26.1 g, 103 mmol) from step 1 is dissolved in THF (900 ml). Manganese dioxide (106.6 g) is added and the mixture is stirred for 2h at room temperature. After the reaction is complete the mixture is filtered through celite and washed with ethyl acetate. The filtrate is concentrated under reduced pressure, dried to give the desired aldehyde (22.9 g, 89%).
- Step 3 This intermediate was prepared from indole, prepared in step 2 above and 2- (bromomethyl)naphthalene, according to the procedure described in step 1, Example 122.
- Step 4 To sodium hydride (0.025 g, 60% oil dispersion, 0.63 mmol) in 7.5 mL of THF is added trimethyl phosphonoacetate (0.1 mL, 0.62 mmol) in 2.5 mL of THF at room temperature. The reaction is stirred for 10 minutes. Next the aldehyde (0.24 g, 0.62 mmol) prepared in step 3 above in 2.5 mL THF is added dropwise at room temperature. Reaction is stirred for another 30 minutes NOT FURNISHED UPON FILING
- Step 1 p-Toluoyl chloride (0.8 M) was added to triethylamine (2.44 eq) and methoxymethyl amine HCl (1.1 eq) dissolved in methylene chloride at 0°C over 20 min. The reaction was allowed to warm to 25°C. After stirring at 25°C for 1 day, workup with methylene chloride and water afforded crude product in ca. 100% yield.
- Step 3 The tolyl ketone from step 2 was dissolved in carbon tetrachloride (0.19M), and
- Step 4 The intermediate from step 3, Example 131 was dissolved in dry DMF (0.1 M), followed by NaH (1.2 eq). After 1.5 h at 25 °C, added the bromobenzyl ketone from step 3 and stirred for 1 d at 25°C. Workup (ethyl acetate/hexanes) and trituration (ethyl acetate/hexanes) afforded the product in 46% yield.
- Step 5 The product from step 4 was dissolved in methylene chloride and 1 N HCl (ca. 0.04 M) and stirred at 25°C for 1 h. Workup (sodium bicarbonate), and trituration with ether afforded the product alcohol (89%).
- Step 6 The alcohol from step 5 was dissolved in dry methylene chloride (0.014 M), treated with thionyl chloride (1.2 eq) and stirred at 25°C for 1 d. Concentration and trituration with ethyl acetate/hexanes afforded the product chloride (100%). NOT FURNISHED UPON FILING
- a typical assay consisted of the lipid mixture (85 ⁇ l) to which was added consecutively, the inhibitor (5 ⁇ l in DMSO) and cPLA, 10 ng for an automated system or 1 ng for a manual assay, in 10 ⁇ l of the BSA buffer. This assay was conducted by either the manual assay or automated assay protocol described below.
- Soluble Substrate Assay (LysoPO l-[ l4 C]-palmitoyl-2-hydroxyphosphotidyl-choline (57 mCi/mmol) (final concentration 4.4 ⁇ M) was dried under a stream of nitrogen. The lipid was resuspended by vortexing 80 mM Hepes pH 7.5, 1 mM EDTA (1.2 x final concentration). A typical assay consisted of lipid suspension (85 ⁇ l) to which was added consecutively the inhibitor (5 ⁇ l in DMSO) and cPLA, 200 ng in 80 mM Hepes pH 7.5, 2 mM DTT and 1 M EDTA. This assay was conducted by either the manual assay or automated assay protocol described below.
- RBL-2H3 cells were routinely cultured as 37C in a 5 % C0 2 atmosphere in minimal essential medium containing nonessential amino acids and 12% fetal calf serum. The day before the experiment, cells were seeded into spinner flasks at 3 x 10 cells/ml and 100 ng/ml DNP specific-IgE was added. After 20 hrs, the cells were harvested by centrifugation and washed once in serum-free minimal essential media, and resuspended to 2 x 10 cells/ml in serum free media.
- the cells were then preincubated with either inhibitor in DMSO (1 % v/v) or DMSO (1 % v/v) for 15 min at 37C followed by stimulation with DNP-BSA (300 ng/ml). After 6 min, the cells were removed by centrifugation, and the supernatant was assayed for PGD ⁇ content in accordance with known methods.
- 7-hydroxycoumarinyl 6-heptenoate was used as a monomeric substrate for cPLA2 as reported previously (Huang, Z. et al., 1994, Analytical Biochemistry 222, 110-115). Inhibitors were mixed with 200 ⁇ L assay buffer (80mM Hepes, pH 7.5, 1 mM EDTA) containing 60 ⁇ M 7-hydroxycoumarinyl 6- heptenoate. The reaction was initiated by adding 4 ⁇ g cPLA2 in 50 ⁇ L assay buffer. Hydrolysis of the 7-hydroxycoumarinyl 6-heptenoate ester was monitored in a fluorometer by exciting at 360 nm and monitoring emission at 460 nm. Enzyme activity is proportional to the increasein emission at 460 nm per minute. In the presence of a cPLA2 inhibitor, the rate of increase is less.
Abstract
Description



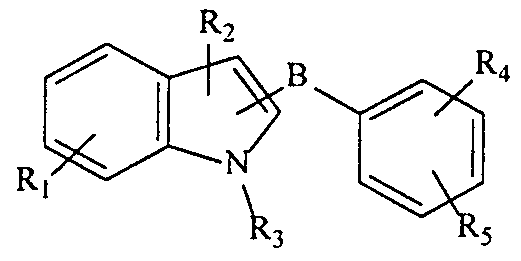





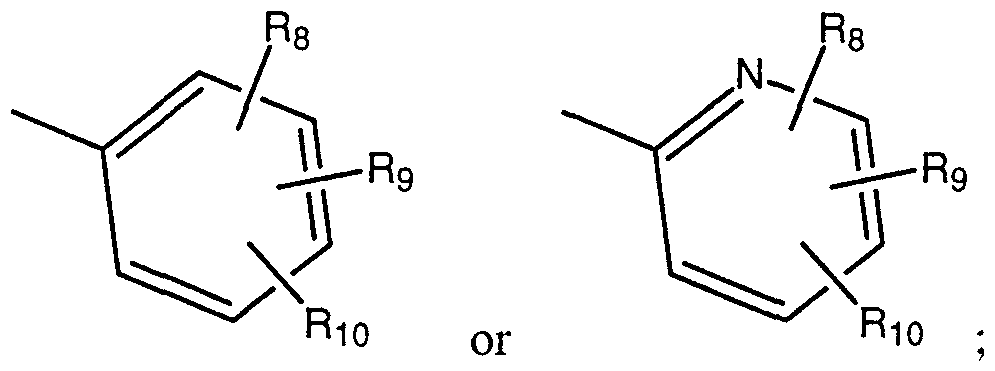
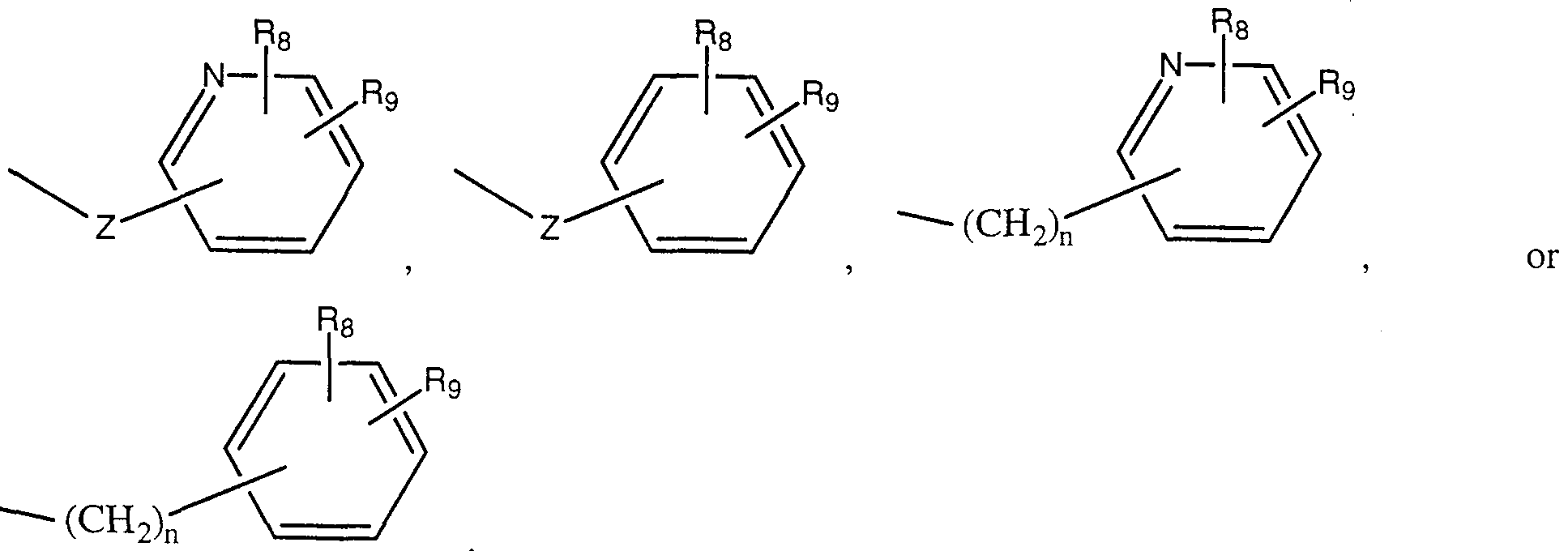























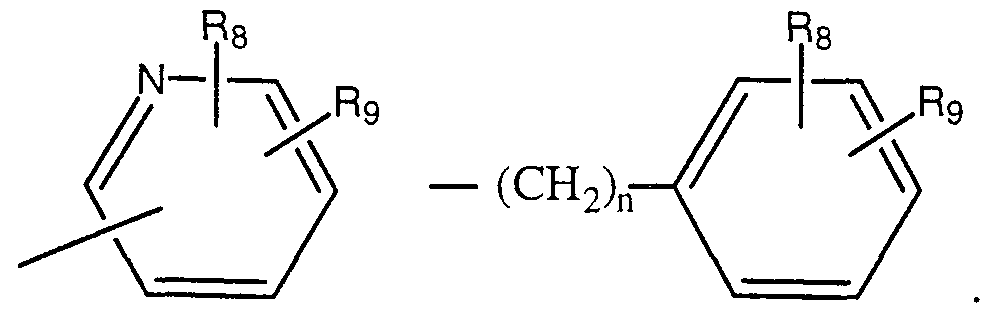
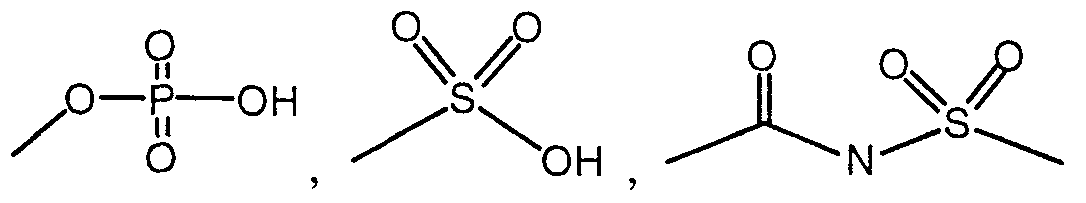



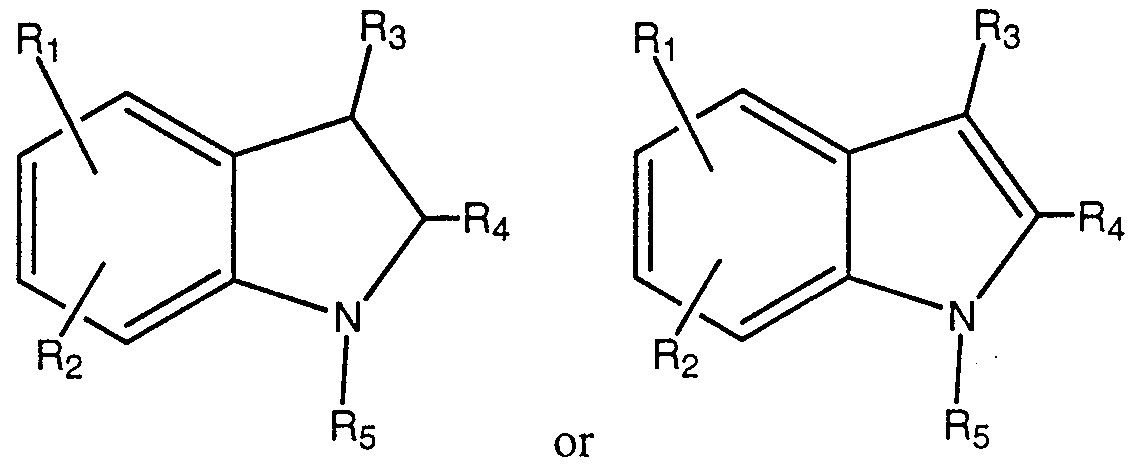
















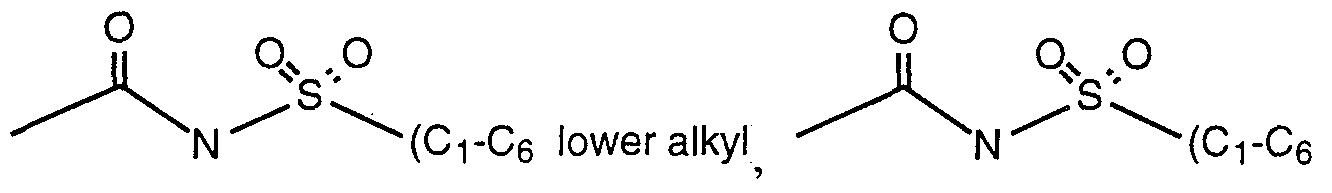

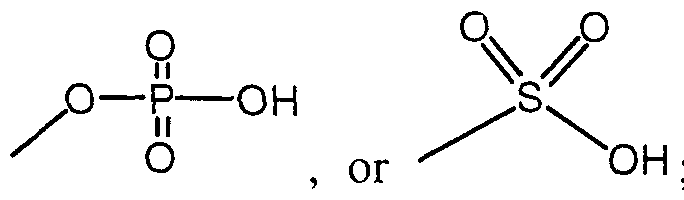



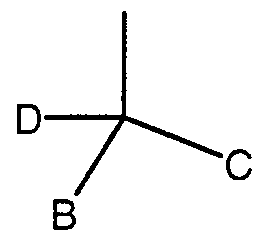
































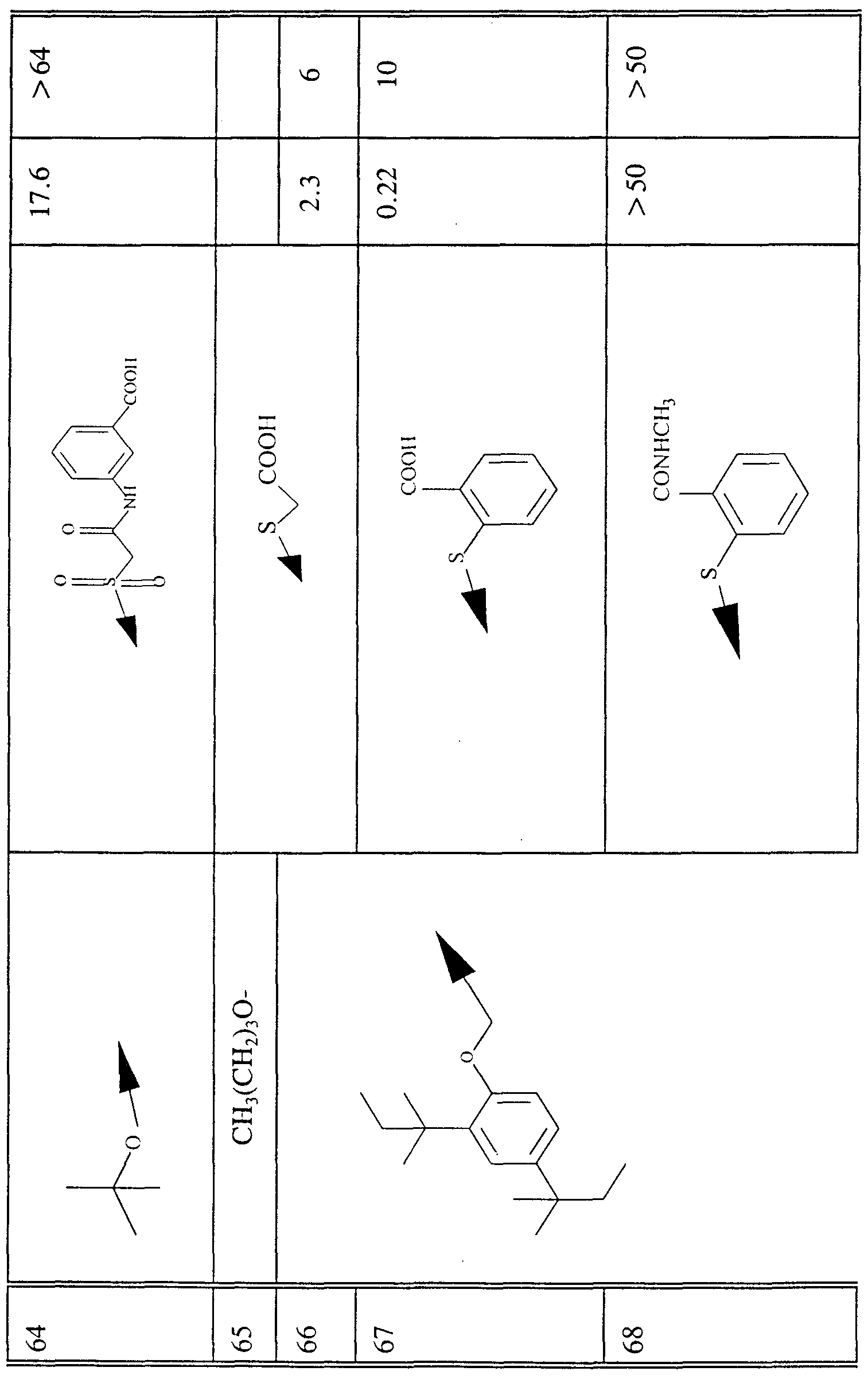














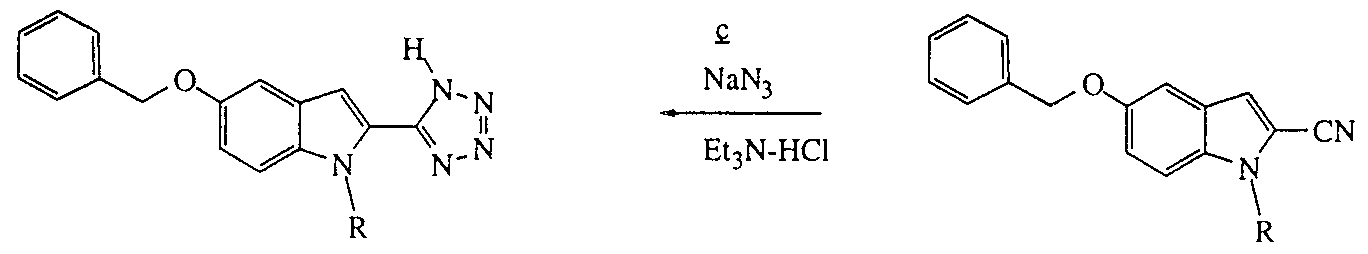
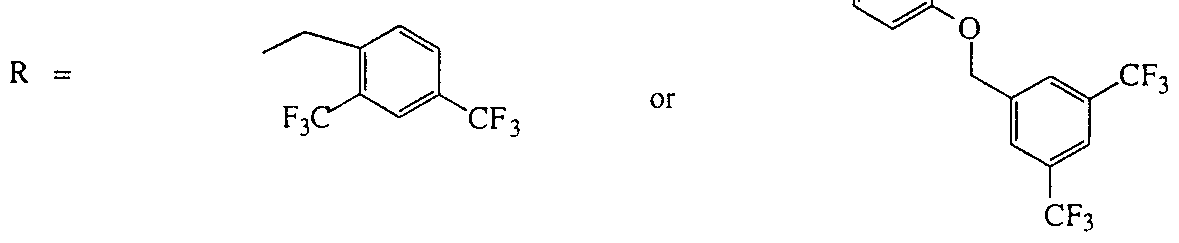



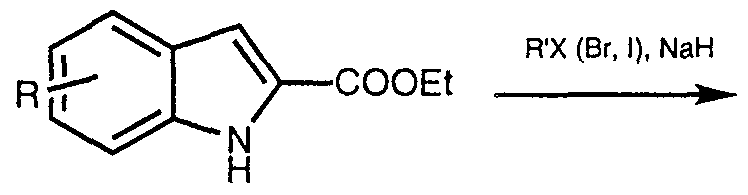


















Claims






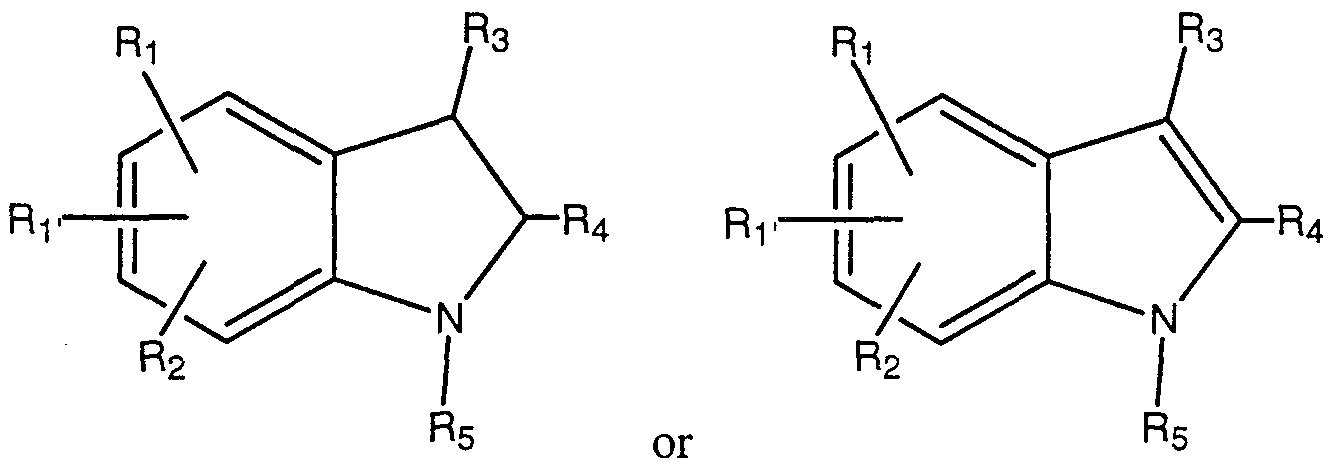































































Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IL13754099A IL137540A0 (en) | 1998-02-25 | 1999-02-17 | Inhibitors of phospholipase a2 |
| EEP200000522A EE200000522A (en) | 1998-02-25 | 1999-02-17 | Phospholipase A2 Enzyme Inhibitor, a Pharmaceutical Composition Containing It and a Method for Inhibiting the Phospholipase Enzyme Activity |
| CA002322163A CA2322163A1 (en) | 1998-02-25 | 1999-02-17 | Inhibitors of phospholipase a2 |
| SK1278-2000A SK12782000A3 (en) | 1998-02-25 | 1999-02-17 | Inhibitors of phospholipase a2 |
| KR1020007009459A KR20010041346A (en) | 1998-02-25 | 1999-02-17 | Inhibitors of phospholipase A2 |
| AU32970/99A AU3297099A (en) | 1998-02-25 | 1999-02-17 | Inhibitors of phospholipase a2 |
| JP2000533428A JP2002504551A (en) | 1998-02-25 | 1999-02-17 | Phospholipase A2 inhibitor |
| HU0100156A HUP0100156A3 (en) | 1998-02-25 | 1999-02-17 | Indole derivatives as inhibitors of phospholipase a2 and use of them for producing pharmaceutical compositions |
| EA200000873A EA200000873A1 (en) | 1998-02-25 | 1999-02-17 | Phospholipase A Inhibitors |
| EP99936073A EP1062216A1 (en) | 1998-02-25 | 1999-02-17 | Inhibitors of phospholipase a2 |
| HR20000513A HRP20000513A2 (en) | 1998-02-25 | 2000-07-31 | Inhibitors of phospholipase a2 |
| NO20004217A NO20004217L (en) | 1998-02-25 | 2000-08-23 | Inhibitors of phospholipase A2 |
| BG104781A BG104781A (en) | 1998-02-25 | 2000-09-19 | Inhibitors of phospholipase a2 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3010298A | 1998-02-25 | 1998-02-25 | |
| US09/030,102 | 1998-02-25 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1999043672A1 true WO1999043672A1 (en) | 1999-09-02 |
| WO1999043672A9 WO1999043672A9 (en) | 2000-05-04 |
Family
ID=21852517
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/003388 WO1999043672A1 (en) | 1998-02-25 | 1999-02-17 | Inhibitors of phospholipase a2 |
Country Status (19)
| Country | Link |
|---|---|
| EP (1) | EP1062216A1 (en) |
| JP (1) | JP2002504551A (en) |
| KR (1) | KR20010041346A (en) |
| CN (1) | CN1298404A (en) |
| AU (1) | AU3297099A (en) |
| BG (1) | BG104781A (en) |
| BR (1) | BR9909242A (en) |
| CA (1) | CA2322163A1 (en) |
| EA (1) | EA200000873A1 (en) |
| EE (1) | EE200000522A (en) |
| HR (1) | HRP20000513A2 (en) |
| HU (1) | HUP0100156A3 (en) |
| ID (1) | ID26123A (en) |
| IL (1) | IL137540A0 (en) |
| NO (1) | NO20004217L (en) |
| PL (1) | PL342516A1 (en) |
| SK (1) | SK12782000A3 (en) |
| TR (1) | TR200002445T2 (en) |
| WO (1) | WO1999043672A1 (en) |
Cited By (114)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001053275A2 (en) * | 2000-01-17 | 2001-07-26 | Bayer Aktiengesellschaft | Substituted aryl ketones |
| WO2002000620A1 (en) * | 2000-06-27 | 2002-01-03 | Smithkline Beecham Corporation | Fatty acid synthase inhibitors |
| WO2002005796A2 (en) * | 2000-07-14 | 2002-01-24 | Eli Lilly And Company | Use of a spla2 inhibitor for the treatment of sepsis |
| WO2002053534A1 (en) * | 2000-12-28 | 2002-07-11 | Daiichi Pharmaceutical Co., Ltd. | Vla-4 inhibitors |
| WO2002094830A2 (en) * | 2001-05-23 | 2002-11-28 | Merck Frosst Canada & Co. | DIHYDROPYRROLO[1,2-A]INDOLE AND TETRAHYDROPYRIDO[1,2-a]-INDOLE DERIVATIVES AS PROSTAGLANDIN D2 RECEPTOR ANTAGONISTS |
| US6525083B2 (en) | 2000-07-25 | 2003-02-25 | Merck & Co., Inc. | N-substituted indoles useful in the treatment of diabetes |
| US6635771B2 (en) | 2001-12-03 | 2003-10-21 | Wyeth | N-benzhydryl indole compounds |
| US6797708B2 (en) | 2001-12-03 | 2004-09-28 | Wyeth | Inhibitors of cytosolic phospholipase A2 |
| US6872744B2 (en) | 2001-09-27 | 2005-03-29 | Syntex (U.S.A.) Llc | Indole derivatives as anti-inflammatory agents |
| US6984735B2 (en) | 2001-12-03 | 2006-01-10 | Wyeth | Process for making an aldehyde |
| EP1633716A2 (en) * | 2003-04-16 | 2006-03-15 | Bristol-Myers Squibb Company | Biarylmethyl indolines, indoles and tetrahydroquinolines, useful as serine protease inhibitors |
| US7022714B2 (en) | 2002-07-31 | 2006-04-04 | Euro-Celtique S.A. | Aryl substituted benzimidazoles and their use as sodium channel blockers |
| US7056943B2 (en) | 2002-12-10 | 2006-06-06 | Wyeth | Substituted indole oxo-acetyl amino acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7074817B2 (en) | 2001-06-20 | 2006-07-11 | Wyeth | Substituted indole acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7078429B2 (en) | 2002-12-10 | 2006-07-18 | Wyeth | Substituted 3-carbonyl-1H-indol-1-yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7078419B2 (en) | 2003-03-10 | 2006-07-18 | Boehringer Ingelheim Pharmaceuticals, Inc. | Cytokine inhibitors |
| WO2006077364A1 (en) * | 2005-01-19 | 2006-07-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| WO2006077366A1 (en) * | 2005-01-19 | 2006-07-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| WO2006077367A1 (en) * | 2005-01-19 | 2006-07-27 | Biolipox Ab | Indoles useful in the treatment of inflamation |
| WO2006077365A1 (en) * | 2005-01-19 | 2006-07-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| US7094790B2 (en) | 2003-05-07 | 2006-08-22 | Abbott Laboratories | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| US7101903B2 (en) | 2002-12-10 | 2006-09-05 | Wyeth | Substituted dihydropyrano indole-3,4-dione derivatives as inhibitiors of plasminogen activator inhibitor-1 (PAI-1) |
| US7101875B2 (en) | 2001-12-03 | 2006-09-05 | Wyeth | Methods for treating arthritic disorders |
| US7141592B2 (en) | 2003-09-25 | 2006-11-28 | Wyeth | Substituted oxadiazolidinediones |
| US7163954B2 (en) | 2003-09-25 | 2007-01-16 | Wyeth | Substituted naphthyl benzothiophene acids |
| EP1747003A1 (en) * | 2004-05-03 | 2007-01-31 | Ilypsa, Inc. | Phospholipase inhibitors localized in the gastrointestinal lumen |
| US7183309B2 (en) | 2004-10-27 | 2007-02-27 | Janssen Pharmaceutica N.V. | Indole derivatives useful as progesterone receptor modulators |
| US7186746B2 (en) | 2002-08-29 | 2007-03-06 | Merck & Co., Inc. | Indoles having anti-diabetic activity |
| WO2007048847A2 (en) * | 2005-10-28 | 2007-05-03 | Nikem Research S.R.L. | Indole and azaindole derivatives for the treatment of inflammatory and autoimmune diseases |
| EP1814865A2 (en) * | 2004-09-21 | 2007-08-08 | Athersys, Inc. | Benzimidazole acetic acids exhibiting crth2 receptor antagonism and uses thereof |
| US7259182B2 (en) | 2002-12-10 | 2007-08-21 | Wyeth | Aryl, aryloxy, and aklyloxy substituted 1H-indol-3-yl glyoxylic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7265148B2 (en) | 2003-09-25 | 2007-09-04 | Wyeth | Substituted pyrrole-indoles |
| US7268159B2 (en) | 2003-09-25 | 2007-09-11 | Wyeth | Substituted indoles |
| US7291639B2 (en) | 2001-06-20 | 2007-11-06 | Wyeth | Aryloxy-acetic acid compounds useful as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| WO2008009924A2 (en) * | 2006-07-18 | 2008-01-24 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| US7332521B2 (en) | 2003-09-25 | 2008-02-19 | Wyeth | Substituted indoles |
| US7342039B2 (en) | 2003-09-25 | 2008-03-11 | Wyeth | Substituted indole oximes |
| US7348351B2 (en) | 2002-12-10 | 2008-03-25 | Wyeth | Substituted 3-alkyl and 3-arylalkyl 1H-indol-1yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7351730B2 (en) | 2001-06-20 | 2008-04-01 | Wyeth | Substituted naphthyl indole derivatives as inhibitors of plasminogen activator inhibitor type-1 (PAI-1) |
| US7351726B2 (en) | 2003-09-25 | 2008-04-01 | Wyeth | Substituted oxadiazolidinediones |
| US7393960B2 (en) | 2002-08-29 | 2008-07-01 | Merck & Co., Inc. | Indoles having anti-diabetic activity |
| US7411083B2 (en) | 2003-09-25 | 2008-08-12 | Wyeth | Substituted acetic acid derivatives |
| US7420083B2 (en) | 2003-09-25 | 2008-09-02 | Wyeth | Substituted aryloximes |
| US7442805B2 (en) | 2003-09-25 | 2008-10-28 | Wyeth | Substituted sulfonamide-indoles |
| US7446201B2 (en) | 2003-09-25 | 2008-11-04 | Wyeth | Substituted heteroaryl benzofuran acids |
| US7528262B2 (en) | 2005-10-14 | 2009-05-05 | Athersys, Inc. | Indole derivatives as histamine 3 receptor inhibitors for the treatment of cognitive and sleep disorders, obesity and other CNS disorders |
| US7557135B2 (en) | 2005-05-27 | 2009-07-07 | Wyeth | Inhibitors of cytosolic phospholipase A2 |
| US7582773B2 (en) | 2003-09-25 | 2009-09-01 | Wyeth | Substituted phenyl indoles |
| US7605156B2 (en) | 2001-12-03 | 2009-10-20 | Wyeth | Methods for the use of inhibitors of cytosolic phospholipase A2 |
| US7666898B2 (en) | 2005-11-03 | 2010-02-23 | Ilypsa, Inc. | Multivalent indole compounds and use thereof as phospholipase-A2 inhibitors |
| US7683091B2 (en) | 2005-08-17 | 2010-03-23 | Wyeth | Substituted indoles and methods of their use |
| US7691894B2 (en) | 2003-07-24 | 2010-04-06 | Daiichi Pharmaceutical Co., Ltd. | Cyclohexanecarboxylic acid compound |
| US7705023B2 (en) | 2004-06-18 | 2010-04-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| US7713964B2 (en) | 2001-12-03 | 2010-05-11 | Wyeth Llc | Methods for treating asthmatic conditions |
| US7754747B2 (en) | 2004-08-23 | 2010-07-13 | Wyeth Llc | Oxazolo-naphthyl acids |
| US7763656B2 (en) * | 2002-07-24 | 2010-07-27 | Ptc Therapeutics, Inc. | Use of Acetylamino benzoic acid compounds for nonsense suppression and the treatment of disease |
| US7795280B2 (en) | 2002-11-07 | 2010-09-14 | N.V. Organon | Indoles useful in the treatment of androgen-receptor related diseases |
| US7812036B2 (en) | 2004-04-23 | 2010-10-12 | N.V. Organon | Androgens |
| US7834037B2 (en) | 2005-11-04 | 2010-11-16 | Amira Pharmaceuticals, Inc. | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US7977359B2 (en) | 2005-11-04 | 2011-07-12 | Amira Pharmaceuticals, Inc. | 5-lipdxygenase-activating protein (FLAP) inhibitors |
| EP2431029A2 (en) | 2002-06-07 | 2012-03-21 | Kieran Francis Scott | Method of inhibiting prostate cancer cell proliferation |
| US8211885B2 (en) | 2008-01-31 | 2012-07-03 | Sanofi-Aventis | Cyclic indole-3-carboxamides, their preparation and their use as pharmaceuticals |
| US8329914B2 (en) | 2008-10-31 | 2012-12-11 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8394969B2 (en) | 2008-09-26 | 2013-03-12 | Merck Sharp & Dohme Corp. | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8399666B2 (en) | 2005-11-04 | 2013-03-19 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US8410284B2 (en) | 2008-10-22 | 2013-04-02 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8563746B2 (en) | 2008-10-29 | 2013-10-22 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8598156B2 (en) | 2010-03-25 | 2013-12-03 | Glaxosmithkline Llc | Chemical compounds |
| US8772495B2 (en) | 2008-05-23 | 2014-07-08 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein inhibitor |
| US8884020B2 (en) | 2006-08-07 | 2014-11-11 | Ironwood Pharmaceuticals, Inc. | Indole compounds |
| US8895596B2 (en) | 2010-02-25 | 2014-11-25 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8952171B2 (en) | 2013-03-15 | 2015-02-10 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9000025B2 (en) | 2010-08-20 | 2015-04-07 | Amira Pharmaceuticals, Inc. | Autotaxin inhibitors and uses thereof |
| US9012450B2 (en) | 2011-12-28 | 2015-04-21 | Global Blood Therapeutics, Inc. | Substituted heteroaryl aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US9018210B2 (en) | 2011-12-28 | 2015-04-28 | Global Blood Therapeutics, Inc. | Substituted benzaldehyde compounds and methods for their use in increasing tissue oxygenation |
| US9062055B2 (en) | 2010-06-21 | 2015-06-23 | Incyte Corporation | Fused pyrrole derivatives as PI3K inhibitors |
| US9260416B2 (en) | 2011-05-27 | 2016-02-16 | Amira Pharmaceuticals, Inc. | Heterocyclic autotaxin inhibitors and uses thereof |
| US9388161B2 (en) | 2013-11-18 | 2016-07-12 | Forma Therapeutics, Inc. | Tetrahydroquinoline compositions as BET bromodomain inhibitors |
| US9422279B2 (en) | 2013-03-15 | 2016-08-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9458139B2 (en) | 2013-03-15 | 2016-10-04 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9556166B2 (en) | 2011-05-12 | 2017-01-31 | Proteostasis Therapeutics, Inc. | Proteostasis regulators |
| US9573958B2 (en) | 2012-08-31 | 2017-02-21 | Principia Biopharma, Inc. | Benzimidazole derivatives as ITK inhibitors |
| US9604999B2 (en) | 2013-03-15 | 2017-03-28 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9657012B2 (en) | 2010-12-22 | 2017-05-23 | Ironwood Pharmaceuticals, Inc. | FAAH inhibitors |
| US9707233B2 (en) | 2011-09-02 | 2017-07-18 | Incyte Holdings Corporation | Heterocyclylamines as PI3K inhibitors |
| US9732097B2 (en) | 2015-05-11 | 2017-08-15 | Incyte Corporation | Process for the synthesis of a phosphoinositide 3-kinase inhibitor |
| US9802900B2 (en) | 2013-03-15 | 2017-10-31 | Global Blood Therapeutics, Inc. | Bicyclic heteroaryl compounds and uses thereof for the modulation of hemoglobin |
| US9815839B2 (en) | 2010-12-20 | 2017-11-14 | Incyte Corporation | N-(1-(substituted-phenyl)ethyl)-9H-purin-6-amines as PI3K inhibitors |
| US9849135B2 (en) | 2013-01-25 | 2017-12-26 | President And Fellows Of Harvard College | USP14 inhibitors for treating or preventing viral infections |
| US9850203B2 (en) | 2013-09-26 | 2017-12-26 | Pharmakea, Inc. | Autotaxin inhibitor compounds |
| US9850262B2 (en) | 2013-11-12 | 2017-12-26 | Proteostasis Therapeutics, Inc. | Proteasome activity enhancing compounds |
| US9944646B2 (en) | 2012-04-02 | 2018-04-17 | Incyte Holdings Corporation | Bicyclic azaheterocyclobenzylamines as PI3K inhibitors |
| US9957250B2 (en) | 2013-03-15 | 2018-05-01 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9975907B2 (en) | 2009-06-29 | 2018-05-22 | Incyte Holdings Corporation | Pyrimidinones as PI3K inhibitors |
| US9981939B2 (en) | 2013-03-15 | 2018-05-29 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9988401B2 (en) | 2015-05-11 | 2018-06-05 | Incyte Corporation | Crystalline forms of a PI3K inhibitor |
| US10004725B2 (en) | 2015-03-30 | 2018-06-26 | Global Blood Therapeutics, Inc. | Methods of treatment |
| US10011566B2 (en) | 2015-12-15 | 2018-07-03 | Astrazeneca Ab | Compounds |
| US10077277B2 (en) | 2014-06-11 | 2018-09-18 | Incyte Corporation | Bicyclic heteroarylaminoalkyl phenyl derivatives as PI3K inhibitors |
| US10077249B2 (en) | 2016-05-12 | 2018-09-18 | Global Blood Therapeutics, Inc. | Process for synthesizing 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)-pyridin-3-yl)methoxy)benzaldehyde |
| US10100043B2 (en) | 2013-03-15 | 2018-10-16 | Global Blood Therapeutics, Inc. | Substituted aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US10137118B2 (en) | 2014-02-07 | 2018-11-27 | Global Blood Therapeutics, Inc. | Crystalline polymorphs of the free base of 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| US10266551B2 (en) | 2013-03-15 | 2019-04-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10336759B2 (en) | 2015-02-27 | 2019-07-02 | Incyte Corporation | Salts and processes of preparing a PI3K inhibitor |
| US10351568B2 (en) | 2010-01-28 | 2019-07-16 | President And Fellows Of Harvard College | Compositions and methods for enhancing proteasome activity |
| US10377769B2 (en) | 2013-11-18 | 2019-08-13 | Forma Therapeutics, Inc. | Benzopiperazine compositions as BET bromodomain inhibitors |
| US10450269B1 (en) | 2013-11-18 | 2019-10-22 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10493035B2 (en) | 2016-10-12 | 2019-12-03 | Global Blood Therapeutics, Inc. | Tablets comprising 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| US11014884B2 (en) | 2018-10-01 | 2021-05-25 | Global Blood Therapeutics, Inc. | Modulators of hemoglobin |
| US11020382B2 (en) | 2015-12-04 | 2021-06-01 | Global Blood Therapeutics, Inc. | Dosing regimens for 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| US11034654B2 (en) | 2017-06-14 | 2021-06-15 | Astrazeneca Ab | 2,3-dihydroisoindole-1-carboxamides useful as ROR-gamma modulators |
| US11053195B2 (en) | 2013-03-15 | 2021-07-06 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US11236109B2 (en) | 2013-03-15 | 2022-02-01 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US11471455B2 (en) | 2018-10-05 | 2022-10-18 | Annapurna Bio, Inc. | Compounds and compositions for treating conditions associated with APJ receptor activity |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5001650B2 (en) * | 2003-07-11 | 2012-08-15 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Benzimidazole carboxamide |
| NZ544970A (en) * | 2003-07-28 | 2009-02-28 | Janssen Pharmaceutica Nv | Benzimidazole, benzthiazole and benzoxazole derivatives and their use as LTA4H modulators |
| US7119214B2 (en) * | 2004-04-13 | 2006-10-10 | Cephalon France | Thio-substituted tricyclic and bicyclic aromatic methanesulfinyl derivatives |
| CN101048382B (en) * | 2004-10-27 | 2010-05-05 | 霍夫曼-拉罗奇有限公司 | New indole or benzimidazole derivatives |
| WO2006109633A1 (en) * | 2005-04-07 | 2006-10-19 | Daiichi Sankyo Company, Limited | Substituted indole compound |
| WO2011075643A1 (en) | 2009-12-18 | 2011-06-23 | Incyte Corporation | Substituted heteroaryl fused derivatives as pi3k inhibitors |
| WO2011130342A1 (en) | 2010-04-14 | 2011-10-20 | Incyte Corporation | FUSED DERIVATIVES AS ΡI3Κδ INHIBITORS |
| WO2012018980A2 (en) * | 2010-08-04 | 2012-02-09 | Yun Michael Shim | Compositions and methods for treating inflammatory diseases |
| US9108984B2 (en) | 2011-03-14 | 2015-08-18 | Incyte Corporation | Substituted diamino-pyrimidine and diamino-pyridine derivatives as PI3K inhibitors |
| US9126948B2 (en) | 2011-03-25 | 2015-09-08 | Incyte Holdings Corporation | Pyrimidine-4,6-diamine derivatives as PI3K inhibitors |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0620215A1 (en) * | 1993-04-16 | 1994-10-19 | Eli Lilly And Company | 1H-indole-3-acetamide derivatives as sPLA2 inhibitors |
| WO1995013266A1 (en) * | 1993-11-12 | 1995-05-18 | Merckle Gmbh Chem.-Pharm. Fabrik | Acylpyrrole-alkanoic acids and indole-2-alkanoic acids plus their derivatives for use as inhibitors of phospholipase a¿2? |
| WO1997005135A1 (en) * | 1995-07-31 | 1997-02-13 | Shionogi & Co., Ltd. | Pyrrolidine derivatives having phospholipase a2 inhibitory activity |
| WO1998005637A1 (en) * | 1996-08-01 | 1998-02-12 | Merckle Gmbh | Acylpyrroldicarboxylic acids and acylindoldicarboxylic acids and their derivatives and inhibitors of the cytosolic phospholipase a¿2? |
| WO1998008818A1 (en) * | 1996-08-26 | 1998-03-05 | Genetics Institute, Inc. | Inhibitors of phospholipase enzymes |
-
1999
- 1999-02-17 IL IL13754099A patent/IL137540A0/en unknown
- 1999-02-17 JP JP2000533428A patent/JP2002504551A/en not_active Withdrawn
- 1999-02-17 WO PCT/US1999/003388 patent/WO1999043672A1/en not_active Application Discontinuation
- 1999-02-17 HU HU0100156A patent/HUP0100156A3/en unknown
- 1999-02-17 EA EA200000873A patent/EA200000873A1/en unknown
- 1999-02-17 TR TR2000/02445T patent/TR200002445T2/en unknown
- 1999-02-17 PL PL99342516A patent/PL342516A1/en unknown
- 1999-02-17 EE EEP200000522A patent/EE200000522A/en unknown
- 1999-02-17 SK SK1278-2000A patent/SK12782000A3/en unknown
- 1999-02-17 EP EP99936073A patent/EP1062216A1/en not_active Withdrawn
- 1999-02-17 CA CA002322163A patent/CA2322163A1/en not_active Abandoned
- 1999-02-17 BR BR9909242-5A patent/BR9909242A/en not_active IP Right Cessation
- 1999-02-17 AU AU32970/99A patent/AU3297099A/en not_active Abandoned
- 1999-02-17 CN CN99805379A patent/CN1298404A/en active Pending
- 1999-02-17 KR KR1020007009459A patent/KR20010041346A/en not_active Application Discontinuation
- 1999-02-17 ID IDW20001568A patent/ID26123A/en unknown
-
2000
- 2000-07-31 HR HR20000513A patent/HRP20000513A2/en not_active Application Discontinuation
- 2000-08-23 NO NO20004217A patent/NO20004217L/en not_active Application Discontinuation
- 2000-09-19 BG BG104781A patent/BG104781A/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0620215A1 (en) * | 1993-04-16 | 1994-10-19 | Eli Lilly And Company | 1H-indole-3-acetamide derivatives as sPLA2 inhibitors |
| WO1995013266A1 (en) * | 1993-11-12 | 1995-05-18 | Merckle Gmbh Chem.-Pharm. Fabrik | Acylpyrrole-alkanoic acids and indole-2-alkanoic acids plus their derivatives for use as inhibitors of phospholipase a¿2? |
| WO1997005135A1 (en) * | 1995-07-31 | 1997-02-13 | Shionogi & Co., Ltd. | Pyrrolidine derivatives having phospholipase a2 inhibitory activity |
| WO1998005637A1 (en) * | 1996-08-01 | 1998-02-12 | Merckle Gmbh | Acylpyrroldicarboxylic acids and acylindoldicarboxylic acids and their derivatives and inhibitors of the cytosolic phospholipase a¿2? |
| WO1998008818A1 (en) * | 1996-08-26 | 1998-03-05 | Genetics Institute, Inc. | Inhibitors of phospholipase enzymes |
Cited By (194)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6864219B2 (en) | 2000-01-17 | 2005-03-08 | Bayer Aktiengesellschaft | Substituted aryl ketones |
| WO2001053275A2 (en) * | 2000-01-17 | 2001-07-26 | Bayer Aktiengesellschaft | Substituted aryl ketones |
| WO2001053275A3 (en) * | 2000-01-17 | 2003-04-17 | Bayer Ag | Substituted aryl ketones |
| WO2002000620A1 (en) * | 2000-06-27 | 2002-01-03 | Smithkline Beecham Corporation | Fatty acid synthase inhibitors |
| US6670388B1 (en) | 2000-06-27 | 2003-12-30 | Smithkline Beecham Corporation | Fatty acid synthase inhibitors |
| WO2002005796A2 (en) * | 2000-07-14 | 2002-01-24 | Eli Lilly And Company | Use of a spla2 inhibitor for the treatment of sepsis |
| WO2002005796A3 (en) * | 2000-07-14 | 2002-09-06 | Lilly Co Eli | Use of a spla2 inhibitor for the treatment of sepsis |
| US6525083B2 (en) | 2000-07-25 | 2003-02-25 | Merck & Co., Inc. | N-substituted indoles useful in the treatment of diabetes |
| US7157487B2 (en) | 2000-12-28 | 2007-01-02 | Daiichi Pharmaceutical Co., Ltd. | Vla-4 inhibitors |
| CN100471838C (en) * | 2000-12-28 | 2009-03-25 | 第一制药株式会社 | VLA-4 inhibitors |
| WO2002053534A1 (en) * | 2000-12-28 | 2002-07-11 | Daiichi Pharmaceutical Co., Ltd. | Vla-4 inhibitors |
| WO2002094830A3 (en) * | 2001-05-23 | 2003-03-06 | Merck Frosst Canada Inc | DIHYDROPYRROLO[1,2-A]INDOLE AND TETRAHYDROPYRIDO[1,2-a]-INDOLE DERIVATIVES AS PROSTAGLANDIN D2 RECEPTOR ANTAGONISTS |
| WO2002094830A2 (en) * | 2001-05-23 | 2002-11-28 | Merck Frosst Canada & Co. | DIHYDROPYRROLO[1,2-A]INDOLE AND TETRAHYDROPYRIDO[1,2-a]-INDOLE DERIVATIVES AS PROSTAGLANDIN D2 RECEPTOR ANTAGONISTS |
| US7291639B2 (en) | 2001-06-20 | 2007-11-06 | Wyeth | Aryloxy-acetic acid compounds useful as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7368471B2 (en) | 2001-06-20 | 2008-05-06 | Wyeth | Substituted indole acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7074817B2 (en) | 2001-06-20 | 2006-07-11 | Wyeth | Substituted indole acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7351730B2 (en) | 2001-06-20 | 2008-04-01 | Wyeth | Substituted naphthyl indole derivatives as inhibitors of plasminogen activator inhibitor type-1 (PAI-1) |
| US7629377B2 (en) | 2001-06-20 | 2009-12-08 | Wyeth | Substituted naphthyl indole derivatives as inhibitors of plasminogen activator inhibitor type-1 (PAI-1) |
| US7410994B2 (en) | 2001-09-27 | 2008-08-12 | Roche Palo Alto Llc | Indole derivatives as anti-inflammatory agents |
| US6872744B2 (en) | 2001-09-27 | 2005-03-29 | Syntex (U.S.A.) Llc | Indole derivatives as anti-inflammatory agents |
| US7074939B2 (en) | 2001-09-27 | 2006-07-11 | Roche Palo Alto Llc | Indole derivatives as anti-inflammatory agents |
| US6984735B2 (en) | 2001-12-03 | 2006-01-10 | Wyeth | Process for making an aldehyde |
| US7101875B2 (en) | 2001-12-03 | 2006-09-05 | Wyeth | Methods for treating arthritic disorders |
| US7713964B2 (en) | 2001-12-03 | 2010-05-11 | Wyeth Llc | Methods for treating asthmatic conditions |
| US7605156B2 (en) | 2001-12-03 | 2009-10-20 | Wyeth | Methods for the use of inhibitors of cytosolic phospholipase A2 |
| US6635771B2 (en) | 2001-12-03 | 2003-10-21 | Wyeth | N-benzhydryl indole compounds |
| US6797708B2 (en) | 2001-12-03 | 2004-09-28 | Wyeth | Inhibitors of cytosolic phospholipase A2 |
| US7906548B2 (en) | 2001-12-03 | 2011-03-15 | Wyeth Llc | Methods for the use of inhibitors of cytosolic phospholipase A2 |
| EP2431029A2 (en) | 2002-06-07 | 2012-03-21 | Kieran Francis Scott | Method of inhibiting prostate cancer cell proliferation |
| US7763656B2 (en) * | 2002-07-24 | 2010-07-27 | Ptc Therapeutics, Inc. | Use of Acetylamino benzoic acid compounds for nonsense suppression and the treatment of disease |
| US7022714B2 (en) | 2002-07-31 | 2006-04-04 | Euro-Celtique S.A. | Aryl substituted benzimidazoles and their use as sodium channel blockers |
| US7393960B2 (en) | 2002-08-29 | 2008-07-01 | Merck & Co., Inc. | Indoles having anti-diabetic activity |
| US7345085B2 (en) | 2002-08-29 | 2008-03-18 | Merck & Co., Inc. | Indoles having anti-diabetic activity |
| US7186746B2 (en) | 2002-08-29 | 2007-03-06 | Merck & Co., Inc. | Indoles having anti-diabetic activity |
| US7795280B2 (en) | 2002-11-07 | 2010-09-14 | N.V. Organon | Indoles useful in the treatment of androgen-receptor related diseases |
| US7674818B2 (en) | 2002-12-10 | 2010-03-09 | Wyeth Llc | Aryl, aryloxy, alkyloxy substituted 1H-indol-3-yl glyoxylic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7348351B2 (en) | 2002-12-10 | 2008-03-25 | Wyeth | Substituted 3-alkyl and 3-arylalkyl 1H-indol-1yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7566791B2 (en) | 2002-12-10 | 2009-07-28 | Wyeth | Substituted 3-carbonyl-1h-indol-1yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7078429B2 (en) | 2002-12-10 | 2006-07-18 | Wyeth | Substituted 3-carbonyl-1H-indol-1-yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7056943B2 (en) | 2002-12-10 | 2006-06-06 | Wyeth | Substituted indole oxo-acetyl amino acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7259182B2 (en) | 2002-12-10 | 2007-08-21 | Wyeth | Aryl, aryloxy, and aklyloxy substituted 1H-indol-3-yl glyoxylic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7160918B2 (en) | 2002-12-10 | 2007-01-09 | Hassan Mahmoud Elokdah | Substituted indole oxo-acetyl amino acetic acid derivatives as inhibitors of plasminogen activator inhibitor (PAI-1) |
| US7459478B2 (en) | 2002-12-10 | 2008-12-02 | Wyeth | Substituted dihydropyrano indole-3,4-dione derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7101903B2 (en) | 2002-12-10 | 2006-09-05 | Wyeth | Substituted dihydropyrano indole-3,4-dione derivatives as inhibitiors of plasminogen activator inhibitor-1 (PAI-1) |
| US7078419B2 (en) | 2003-03-10 | 2006-07-18 | Boehringer Ingelheim Pharmaceuticals, Inc. | Cytokine inhibitors |
| EP1633716A2 (en) * | 2003-04-16 | 2006-03-15 | Bristol-Myers Squibb Company | Biarylmethyl indolines, indoles and tetrahydroquinolines, useful as serine protease inhibitors |
| EP1633716A4 (en) * | 2003-04-16 | 2008-03-26 | Bristol Myers Squibb Co | Biarylmethyl indolines, indoles and tetrahydroquinolines, useful as serine protease inhibitors |
| US7094790B2 (en) | 2003-05-07 | 2006-08-22 | Abbott Laboratories | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| US8106088B2 (en) | 2003-05-07 | 2012-01-31 | Abbott Laboratories | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| US7358263B2 (en) | 2003-05-07 | 2008-04-15 | Abbott Laboratories | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| US7807697B2 (en) | 2003-05-07 | 2010-10-05 | Abbott Laboratories | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| US7893279B2 (en) | 2003-07-24 | 2011-02-22 | Daiichi Pharmaceutical Co., Ltd. | Cyclohexanecarboxylic acid compound |
| US7691894B2 (en) | 2003-07-24 | 2010-04-06 | Daiichi Pharmaceutical Co., Ltd. | Cyclohexanecarboxylic acid compound |
| US7265148B2 (en) | 2003-09-25 | 2007-09-04 | Wyeth | Substituted pyrrole-indoles |
| US7420083B2 (en) | 2003-09-25 | 2008-09-02 | Wyeth | Substituted aryloximes |
| US7803835B2 (en) | 2003-09-25 | 2010-09-28 | Wyeth Llc | Substituted acetic acid derivatives |
| US7342039B2 (en) | 2003-09-25 | 2008-03-11 | Wyeth | Substituted indole oximes |
| US7351726B2 (en) | 2003-09-25 | 2008-04-01 | Wyeth | Substituted oxadiazolidinediones |
| US7332521B2 (en) | 2003-09-25 | 2008-02-19 | Wyeth | Substituted indoles |
| US7141592B2 (en) | 2003-09-25 | 2006-11-28 | Wyeth | Substituted oxadiazolidinediones |
| US7268159B2 (en) | 2003-09-25 | 2007-09-11 | Wyeth | Substituted indoles |
| US7163954B2 (en) | 2003-09-25 | 2007-01-16 | Wyeth | Substituted naphthyl benzothiophene acids |
| US7411083B2 (en) | 2003-09-25 | 2008-08-12 | Wyeth | Substituted acetic acid derivatives |
| US7582773B2 (en) | 2003-09-25 | 2009-09-01 | Wyeth | Substituted phenyl indoles |
| US7442805B2 (en) | 2003-09-25 | 2008-10-28 | Wyeth | Substituted sulfonamide-indoles |
| US7446201B2 (en) | 2003-09-25 | 2008-11-04 | Wyeth | Substituted heteroaryl benzofuran acids |
| US7812036B2 (en) | 2004-04-23 | 2010-10-12 | N.V. Organon | Androgens |
| EP1750730A4 (en) * | 2004-05-03 | 2008-01-09 | Ilypsa Inc | Treatment of hypercholesterolemia, hypertriglyceridemia and cardiovascular-related conditions using phosphalipase-a2 inhibitors |
| EP1750699A4 (en) * | 2004-05-03 | 2008-01-09 | Ilypsa Inc | Treatment of diet-related conditions using phospholipase-a2 inhibitors comprising indoles and related compounds |
| EP1750699A2 (en) * | 2004-05-03 | 2007-02-14 | Ilypsa, Inc. | Treatment of diet-related conditions using phospholipase-a2 inhibitors comprising indoles and related compounds |
| EP1750730A2 (en) * | 2004-05-03 | 2007-02-14 | Ilypsa, Inc. | Treatment of hypercholesterolemia, hypertriglyceridemia and cardiovascular-related conditions using phosphalipase-a2 inhibitors |
| EP1747003A1 (en) * | 2004-05-03 | 2007-01-31 | Ilypsa, Inc. | Phospholipase inhibitors localized in the gastrointestinal lumen |
| EP1747003A4 (en) * | 2004-05-03 | 2008-01-09 | Ilypsa Inc | Phospholipase inhibitors localized in the gastrointestinal lumen |
| US7705023B2 (en) | 2004-06-18 | 2010-04-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| US7754747B2 (en) | 2004-08-23 | 2010-07-13 | Wyeth Llc | Oxazolo-naphthyl acids |
| EP1814865A4 (en) * | 2004-09-21 | 2009-09-02 | Wyeth Corp | Benzimidazole acetic acids exhibiting crth2 receptor antagonism and uses thereof |
| EP1814865A2 (en) * | 2004-09-21 | 2007-08-08 | Athersys, Inc. | Benzimidazole acetic acids exhibiting crth2 receptor antagonism and uses thereof |
| US7345084B2 (en) | 2004-10-27 | 2008-03-18 | Janssen Pharmaceutica N.V. | Indole derivatives useful as progesterone receptor modulators |
| US7183309B2 (en) | 2004-10-27 | 2007-02-27 | Janssen Pharmaceutica N.V. | Indole derivatives useful as progesterone receptor modulators |
| WO2006077365A1 (en) * | 2005-01-19 | 2006-07-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| WO2006077367A1 (en) * | 2005-01-19 | 2006-07-27 | Biolipox Ab | Indoles useful in the treatment of inflamation |
| WO2006077366A1 (en) * | 2005-01-19 | 2006-07-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| WO2006077364A1 (en) * | 2005-01-19 | 2006-07-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| US8097623B2 (en) | 2005-01-19 | 2012-01-17 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| US8283373B2 (en) | 2005-05-27 | 2012-10-09 | Pfizer Inc. | Inhibitors of cytosolic phospholipase A2 |
| US7557135B2 (en) | 2005-05-27 | 2009-07-07 | Wyeth | Inhibitors of cytosolic phospholipase A2 |
| US7683091B2 (en) | 2005-08-17 | 2010-03-23 | Wyeth | Substituted indoles and methods of their use |
| US8129424B2 (en) | 2005-10-14 | 2012-03-06 | Athersys, Inc. | Indole derivatives as histamine 3 receptor inhibitors for the treatment of cognitive and sleep disorders, obesity and other CNS disorders |
| US7855224B2 (en) | 2005-10-14 | 2010-12-21 | Athersys, Inc. | Indole derivatives as histamine 3 receptor inhibitors for the treatment of cognitive and sleep disorders, obesity and other CNS disorders |
| US7528262B2 (en) | 2005-10-14 | 2009-05-05 | Athersys, Inc. | Indole derivatives as histamine 3 receptor inhibitors for the treatment of cognitive and sleep disorders, obesity and other CNS disorders |
| WO2007048847A3 (en) * | 2005-10-28 | 2007-06-14 | Nikem Research Srl | Indole and azaindole derivatives for the treatment of inflammatory and autoimmune diseases |
| WO2007048847A2 (en) * | 2005-10-28 | 2007-05-03 | Nikem Research S.R.L. | Indole and azaindole derivatives for the treatment of inflammatory and autoimmune diseases |
| US7666898B2 (en) | 2005-11-03 | 2010-02-23 | Ilypsa, Inc. | Multivalent indole compounds and use thereof as phospholipase-A2 inhibitors |
| US7834037B2 (en) | 2005-11-04 | 2010-11-16 | Amira Pharmaceuticals, Inc. | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US7977359B2 (en) | 2005-11-04 | 2011-07-12 | Amira Pharmaceuticals, Inc. | 5-lipdxygenase-activating protein (FLAP) inhibitors |
| US8841295B2 (en) | 2005-11-04 | 2014-09-23 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US8710081B2 (en) | 2005-11-04 | 2014-04-29 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US8399666B2 (en) | 2005-11-04 | 2013-03-19 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| WO2008009924A3 (en) * | 2006-07-18 | 2008-03-27 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| WO2008009924A2 (en) * | 2006-07-18 | 2008-01-24 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| US8884020B2 (en) | 2006-08-07 | 2014-11-11 | Ironwood Pharmaceuticals, Inc. | Indole compounds |
| US8211885B2 (en) | 2008-01-31 | 2012-07-03 | Sanofi-Aventis | Cyclic indole-3-carboxamides, their preparation and their use as pharmaceuticals |
| US8772495B2 (en) | 2008-05-23 | 2014-07-08 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein inhibitor |
| US8394969B2 (en) | 2008-09-26 | 2013-03-12 | Merck Sharp & Dohme Corp. | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8410284B2 (en) | 2008-10-22 | 2013-04-02 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8563746B2 (en) | 2008-10-29 | 2013-10-22 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8329914B2 (en) | 2008-10-31 | 2012-12-11 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US10428087B2 (en) | 2009-06-29 | 2019-10-01 | Incyte Corporation | Pyrimidinones as PI3K inhibitors |
| US10829502B2 (en) | 2009-06-29 | 2020-11-10 | Incyte Corporation | Pyrimidinones as PI3K inhibitors |
| US9975907B2 (en) | 2009-06-29 | 2018-05-22 | Incyte Holdings Corporation | Pyrimidinones as PI3K inhibitors |
| US11401280B2 (en) | 2009-06-29 | 2022-08-02 | Incyte Holdings Corporation | Pyrimidinones as PI3K inhibitors |
| US10351568B2 (en) | 2010-01-28 | 2019-07-16 | President And Fellows Of Harvard College | Compositions and methods for enhancing proteasome activity |
| US8895596B2 (en) | 2010-02-25 | 2014-11-25 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8598156B2 (en) | 2010-03-25 | 2013-12-03 | Glaxosmithkline Llc | Chemical compounds |
| US9062055B2 (en) | 2010-06-21 | 2015-06-23 | Incyte Corporation | Fused pyrrole derivatives as PI3K inhibitors |
| US9849109B2 (en) | 2010-08-20 | 2017-12-26 | Amira Pharmaceuticals, Inc. | Autotaxin inhibitors and uses thereof |
| US9000025B2 (en) | 2010-08-20 | 2015-04-07 | Amira Pharmaceuticals, Inc. | Autotaxin inhibitors and uses thereof |
| US9815839B2 (en) | 2010-12-20 | 2017-11-14 | Incyte Corporation | N-(1-(substituted-phenyl)ethyl)-9H-purin-6-amines as PI3K inhibitors |
| US9657012B2 (en) | 2010-12-22 | 2017-05-23 | Ironwood Pharmaceuticals, Inc. | FAAH inhibitors |
| US9556166B2 (en) | 2011-05-12 | 2017-01-31 | Proteostasis Therapeutics, Inc. | Proteostasis regulators |
| US10532996B2 (en) | 2011-05-12 | 2020-01-14 | Proteostasis Therapeutics, Inc. | Proteostasis regulators |
| US9260416B2 (en) | 2011-05-27 | 2016-02-16 | Amira Pharmaceuticals, Inc. | Heterocyclic autotaxin inhibitors and uses thereof |
| US10646492B2 (en) | 2011-09-02 | 2020-05-12 | Incyte Corporation | Heterocyclylamines as PI3K inhibitors |
| US11819505B2 (en) | 2011-09-02 | 2023-11-21 | Incyte Corporation | Heterocyclylamines as PI3K inhibitors |
| US10092570B2 (en) | 2011-09-02 | 2018-10-09 | Incyte Holdings Corporation | Heterocyclylamines as PI3K inhibitors |
| US9707233B2 (en) | 2011-09-02 | 2017-07-18 | Incyte Holdings Corporation | Heterocyclylamines as PI3K inhibitors |
| US9730939B2 (en) | 2011-09-02 | 2017-08-15 | Incyte Holdings Corporation | Heterocyclylamines as PI3K inhibitors |
| US10376513B2 (en) | 2011-09-02 | 2019-08-13 | Incyte Holdings Corporation | Heterocyclylamines as PI3K inhibitors |
| US11433071B2 (en) | 2011-09-02 | 2022-09-06 | Incyte Corporation | Heterocyclylamines as PI3K inhibitors |
| US9018210B2 (en) | 2011-12-28 | 2015-04-28 | Global Blood Therapeutics, Inc. | Substituted benzaldehyde compounds and methods for their use in increasing tissue oxygenation |
| US10377741B2 (en) | 2011-12-28 | 2019-08-13 | Global Blood Therapeutics, Inc. | Substituted heteroaryl aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US9012450B2 (en) | 2011-12-28 | 2015-04-21 | Global Blood Therapeutics, Inc. | Substituted heteroaryl aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US10822326B2 (en) | 2011-12-28 | 2020-11-03 | Global Blood Therapeutics, Inc. | Substituted heteroaryl aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US10806733B2 (en) | 2011-12-28 | 2020-10-20 | Global Blood Therapeutics, Inc. | Substituted benzaldehyde compounds and methods for their use in increasing tissue oxygenation |
| US10034879B2 (en) | 2011-12-28 | 2018-07-31 | Global Blood Therapeutics, Inc. | Substituted benzaldehyde compounds and methods for their use in increasing tissue oxygenation |
| US9944646B2 (en) | 2012-04-02 | 2018-04-17 | Incyte Holdings Corporation | Bicyclic azaheterocyclobenzylamines as PI3K inhibitors |
| US10259818B2 (en) | 2012-04-02 | 2019-04-16 | Incyte Corporation | Bicyclic azaheterocyclobenzylamines as PI3K inhibitors |
| US9573958B2 (en) | 2012-08-31 | 2017-02-21 | Principia Biopharma, Inc. | Benzimidazole derivatives as ITK inhibitors |
| US9849135B2 (en) | 2013-01-25 | 2017-12-26 | President And Fellows Of Harvard College | USP14 inhibitors for treating or preventing viral infections |
| US10017491B2 (en) | 2013-03-15 | 2018-07-10 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10315991B2 (en) | 2013-03-15 | 2019-06-11 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US8952171B2 (en) | 2013-03-15 | 2015-02-10 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US11530191B2 (en) | 2013-03-15 | 2022-12-20 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9422279B2 (en) | 2013-03-15 | 2016-08-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10100043B2 (en) | 2013-03-15 | 2018-10-16 | Global Blood Therapeutics, Inc. | Substituted aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US10100040B2 (en) | 2013-03-15 | 2018-10-16 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9776960B2 (en) | 2013-03-15 | 2017-10-03 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US11236109B2 (en) | 2013-03-15 | 2022-02-01 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US11053195B2 (en) | 2013-03-15 | 2021-07-06 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10266551B2 (en) | 2013-03-15 | 2019-04-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9802900B2 (en) | 2013-03-15 | 2017-10-31 | Global Blood Therapeutics, Inc. | Bicyclic heteroaryl compounds and uses thereof for the modulation of hemoglobin |
| US10858317B2 (en) | 2013-03-15 | 2020-12-08 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10829470B2 (en) | 2013-03-15 | 2020-11-10 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9981939B2 (en) | 2013-03-15 | 2018-05-29 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9957250B2 (en) | 2013-03-15 | 2018-05-01 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9458139B2 (en) | 2013-03-15 | 2016-10-04 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9604999B2 (en) | 2013-03-15 | 2017-03-28 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10435393B2 (en) | 2013-03-15 | 2019-10-08 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9850203B2 (en) | 2013-09-26 | 2017-12-26 | Pharmakea, Inc. | Autotaxin inhibitor compounds |
| US9850262B2 (en) | 2013-11-12 | 2017-12-26 | Proteostasis Therapeutics, Inc. | Proteasome activity enhancing compounds |
| US11242361B2 (en) | 2013-11-12 | 2022-02-08 | Proteostasis Therapeutics, Inc. | Proteasome activity enhancing compounds |
| US10611750B2 (en) | 2013-11-18 | 2020-04-07 | Forma Therapeutics, Inc. | Tetrahydroquinoline compositions as bet bromodomain inhibitors |
| US10703764B2 (en) | 2013-11-18 | 2020-07-07 | Forma Therapeutics, Inc. | Benzopiperazine compositions as BET bromodomain inhibitors |
| US10336722B2 (en) | 2013-11-18 | 2019-07-02 | Forma Therapeutics, Inc. | Tetrahydroquinoline compositions as BET bromodomain inhibitors |
| US10377769B2 (en) | 2013-11-18 | 2019-08-13 | Forma Therapeutics, Inc. | Benzopiperazine compositions as BET bromodomain inhibitors |
| US9388161B2 (en) | 2013-11-18 | 2016-07-12 | Forma Therapeutics, Inc. | Tetrahydroquinoline compositions as BET bromodomain inhibitors |
| US10450269B1 (en) | 2013-11-18 | 2019-10-22 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US11084831B1 (en) | 2013-11-18 | 2021-08-10 | Forma Therapeutics, Inc. | Benzopiperazine compositions as BET bromodomain inhibitors |
| US11111229B2 (en) | 2013-11-18 | 2021-09-07 | Forma Therapeutics, Inc. | Tetrahydroquinoline compositions as BET bromodomain inhibitors |
| US10722502B2 (en) | 2014-02-07 | 2020-07-28 | Global Blood Therapeutics, Inc. | Crystalline polymorphs of the free base of 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| US10137118B2 (en) | 2014-02-07 | 2018-11-27 | Global Blood Therapeutics, Inc. | Crystalline polymorphs of the free base of 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| US11452720B2 (en) | 2014-02-07 | 2022-09-27 | Global Blood Therapeutics, Inc. | Crystalline polymorphs of the free base of 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| US10479803B2 (en) | 2014-06-11 | 2019-11-19 | Incyte Corporation | Bicyclic heteroarylaminoalkyl phenyl derivatives as PI3K inhibitors |
| US10077277B2 (en) | 2014-06-11 | 2018-09-18 | Incyte Corporation | Bicyclic heteroarylaminoalkyl phenyl derivatives as PI3K inhibitors |
| US11130767B2 (en) | 2014-06-11 | 2021-09-28 | Incyte Corporation | Bicyclic heteroarylaminoalkyl phenyl derivatives as PI3K inhibitors |
| US10336759B2 (en) | 2015-02-27 | 2019-07-02 | Incyte Corporation | Salts and processes of preparing a PI3K inhibitor |
| US11084822B2 (en) | 2015-02-27 | 2021-08-10 | Incyte Corporation | Salts and processes of preparing a PI3K inhibitor |
| US10004725B2 (en) | 2015-03-30 | 2018-06-26 | Global Blood Therapeutics, Inc. | Methods of treatment |
| US10695330B2 (en) | 2015-03-30 | 2020-06-30 | Global Blood Therapeutics, Inc. | Methods of treatment |
| US9732097B2 (en) | 2015-05-11 | 2017-08-15 | Incyte Corporation | Process for the synthesis of a phosphoinositide 3-kinase inhibitor |
| US9988401B2 (en) | 2015-05-11 | 2018-06-05 | Incyte Corporation | Crystalline forms of a PI3K inhibitor |
| US10125150B2 (en) | 2015-05-11 | 2018-11-13 | Incyte Corporation | Crystalline forms of a PI3K inhibitor |
| US11020382B2 (en) | 2015-12-04 | 2021-06-01 | Global Blood Therapeutics, Inc. | Dosing regimens for 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| US10011566B2 (en) | 2015-12-15 | 2018-07-03 | Astrazeneca Ab | Compounds |
| US11453644B1 (en) | 2015-12-15 | 2022-09-27 | Astrazeneca, Ab | Compounds |
| US10988445B2 (en) | 2015-12-15 | 2021-04-27 | Astrazeneca Ab | Compounds |
| US10526286B2 (en) | 2015-12-15 | 2020-01-07 | Astrazeneca Ab | Compounds |
| US10577345B2 (en) | 2016-05-12 | 2020-03-03 | Global Blood Therapeutics, Inc. | Process for synthesizing 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)-pyridin-3-yl)methoxy)benzaldehyde |
| US10077249B2 (en) | 2016-05-12 | 2018-09-18 | Global Blood Therapeutics, Inc. | Process for synthesizing 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)-pyridin-3-yl)methoxy)benzaldehyde |
| US10493035B2 (en) | 2016-10-12 | 2019-12-03 | Global Blood Therapeutics, Inc. | Tablets comprising 2-hydroxy-6-((2-(1-isopropyl-1H-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| US11034654B2 (en) | 2017-06-14 | 2021-06-15 | Astrazeneca Ab | 2,3-dihydroisoindole-1-carboxamides useful as ROR-gamma modulators |
| US11014884B2 (en) | 2018-10-01 | 2021-05-25 | Global Blood Therapeutics, Inc. | Modulators of hemoglobin |
| US11471455B2 (en) | 2018-10-05 | 2022-10-18 | Annapurna Bio, Inc. | Compounds and compositions for treating conditions associated with APJ receptor activity |
| US11944622B2 (en) | 2018-10-05 | 2024-04-02 | Annapurna Bio, Inc. | Compounds and compositions for treating conditions associated with APJ receptor activity |
Also Published As
| Publication number | Publication date |
|---|---|
| TR200002445T2 (en) | 2000-12-21 |
| KR20010041346A (en) | 2001-05-15 |
| CN1298404A (en) | 2001-06-06 |
| EP1062216A1 (en) | 2000-12-27 |
| JP2002504551A (en) | 2002-02-12 |
| EE200000522A (en) | 2002-02-15 |
| BR9909242A (en) | 2000-11-14 |
| HUP0100156A2 (en) | 2001-07-30 |
| IL137540A0 (en) | 2001-07-24 |
| ID26123A (en) | 2000-11-23 |
| SK12782000A3 (en) | 2001-04-09 |
| NO20004217L (en) | 2000-10-23 |
| HUP0100156A3 (en) | 2002-12-28 |
| WO1999043672A9 (en) | 2000-05-04 |
| EA200000873A1 (en) | 2001-04-23 |
| NO20004217D0 (en) | 2000-08-23 |
| CA2322163A1 (en) | 1999-09-02 |
| BG104781A (en) | 2001-10-31 |
| PL342516A1 (en) | 2001-06-18 |
| AU3297099A (en) | 1999-09-15 |
| HRP20000513A2 (en) | 2001-12-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1999043672A1 (en) | Inhibitors of phospholipase a2 | |
| US6630496B1 (en) | Inhibitors of phospholipase enzymes | |
| AU717430B2 (en) | Inhibitors of phospholipase enzymes | |
| AU765427B2 (en) | Inhibitors of phospholipase enzymes | |
| US6828344B1 (en) | Inhibitors of phospholipase enzymes | |
| AU2782699A (en) | Inhibitors of phospholipase enzymes | |
| EP0275667B1 (en) | 3-hetero-substituted-n-benzyl-indoles | |
| US6916841B2 (en) | Inhibitors of phospholipase enzymes | |
| EP1255751B1 (en) | 1,3-dihydro-2h-indol-2-one derivatives and their use as ligands for v1b or v1b and v1a arginine-vasopressin receptors | |
| JPH0680666A (en) | Angiotensin ii antagonist | |
| CZ89394A3 (en) | Derivative of +h-indole-3-acetamide, process of its preparation, and pharmaceutical preparation in which it is comprised | |
| CZ20003117A3 (en) | Phospholipase inhibitors | |
| WO2002006272A1 (en) | Polyfluoroalkyltriazole derivatives, preparation and therapeutic use thereof | |
| MXPA00008294A (en) | Inhibitors of phospholipase a2 | |
| MXPA99001864A (en) | Inhibitors of phospholipase enzymes | |
| EP2640724A1 (en) | Novel indolizine derivatives, and preparation and therapeutic use thereof | |
| MXPA00008295A (en) | Inhibitors of phospholipase enzymes | |
| CZ20003113A3 (en) | Phospholipase inhibitors | |
| MXPA00008292A (en) | Inhibitors of phospholipase enzymes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 99805379.1 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: C2 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: C2 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| COP | Corrected version of pamphlet |
Free format text: PAGES 1-178, DESCRIPTION, REPLACED BY NEW PAGES 1-169; PAGES 179-218, CLAIMS, REPLACED BY NEW PAGES 170-208; DUE TO LATE TRANSMITTAL BY THE RECEIVING OFFICE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 137540 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20000513A Country of ref document: HR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200000704 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999936073 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000/02445 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 506512 Country of ref document: NZ Ref document number: 12782000 Country of ref document: SK |
|
| ENP | Entry into the national phase |
Ref document number: 2322163 Country of ref document: CA Ref document number: 2322163 Country of ref document: CA Kind code of ref document: A Ref document number: 2000 533428 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 32970/99 Country of ref document: AU Ref document number: PA/a/2000/008294 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007009459 Country of ref document: KR Ref document number: PV2000-3117 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200000873 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999936073 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2000-3117 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007009459 Country of ref document: KR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999936073 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1020007009459 Country of ref document: KR |